Alagille症候群(AGS)
<診断のポイント>
- 典型例では乳児期に閉塞性黄疸を生じる。直接ビリルビン高値の乳児が入院してきたら、まず心雑音・椎体や眼球の異常などAGSに特徴的な身体所見がないか、意識してよく調べる。特徴的な顔貌は、幼児期以降に明らかになってくることが多い。
- AGSでは、時に肝外胆管の狭小化や閉塞を伴うことがある。 灰白色便など高度の胆汁うっ滞を呈する乳児では、上記の身体所見を良くチェックして、胆道閉鎖症の手術(肝門部空腸吻合術)を避けることが大切である。
- 肝生検による小葉間胆管の減少の確認が確定診断に有用である。ただし、経皮的肝生検では判定困難な場合があります。また、乳児期早期には特徴的な病理所見が得られないことがあり、注意が必要である。
- AGSは症例毎に症状や重症度が大きく異なるのが特徴であり、非典型例では遺伝子診断が役立つ。
<総論および病態>(図1)
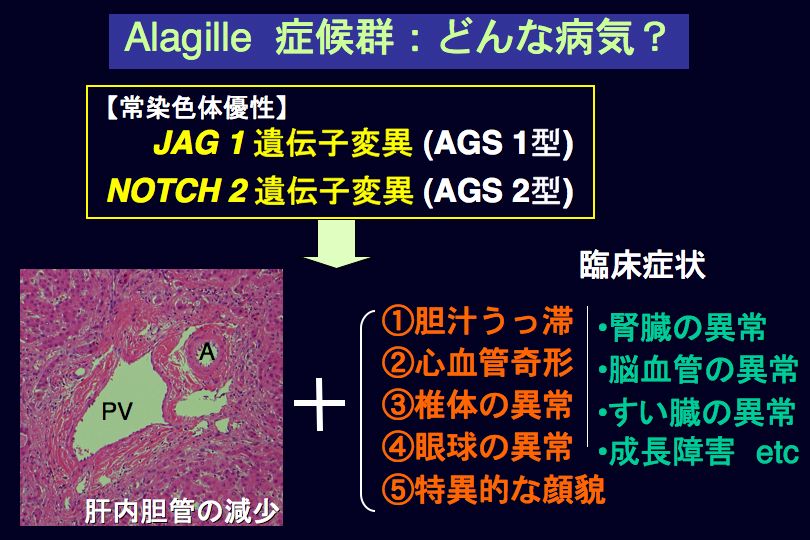
[図1] ※クリックで拡大できます
AGSの病因遺伝子にはいずれもNOTCHシグナル伝達系に属するJAG1遺伝子とNOTCH2遺伝子があり、前者に変異がある場合を1型(AGS1)、後者の遺伝子変異を2型(AGS2)といいます。臨床的に診断されたAGSのうち94%がAGS1によるとする報告があります。いずれの遺伝子も、発生段階における細胞間相互作用を介して、多種類の細胞の分化を制御しており、このためAGSでは多臓器の異常が特徴的な所見となっています。
<診断の詳細>

[図2] ※クリックで拡大できます
◆典型例における診断の進め方を図3に示します。頻度の多い症状を図4に示します。以下に各症状毎に診断の要点を記します。
 [図3] ※クリックで拡大できます |
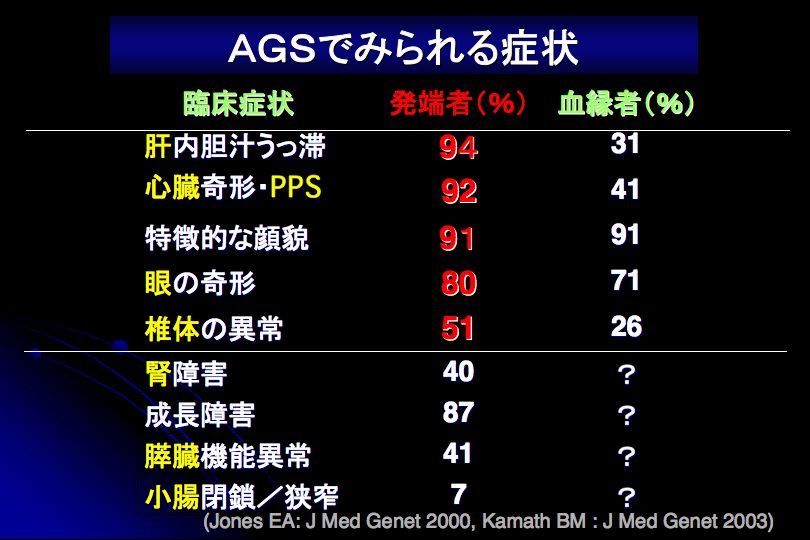 [図4] ※クリックで拡大できます |
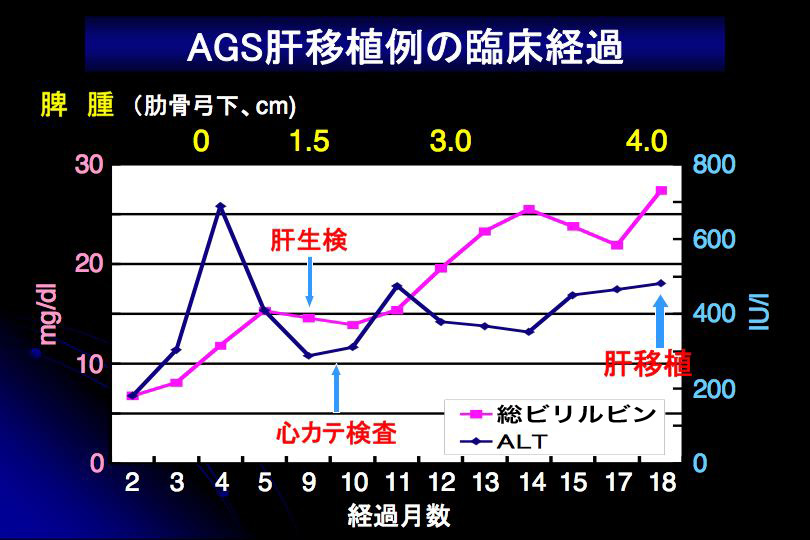
[図5] ※クリックで拡大できます
(1)胆汁うっ滞:黄疸の他に掻痒感、脂溶性ビタミンの不足による出血傾向やくる病がみられることがあります。検査所見上は直接型ビリルビン、γGTP、ALP、血清リポプロテインXの上昇を伴い、胆道閉鎖症と鑑別困難な症例もあるので、術前にAGSに特徴的な臨床症状を探すことが重要です。臨床症状からAGSが強く疑われる場合は、肝生検を生後6か月以降に行うと良いでしょう(図5)。小葉間胆管の減少を確定するためには、少なくとも6個以上の門脈域を評価する必要があり、経皮的肝生検では判定困難な場合も想定されるので多数の検体を採取したり、開放性肝生検を行う場合もあります。◆遺伝子診断の適応として考えられる事項を図3に記しました。
(2)心血管系:特徴的なのは末梢性肺動脈狭窄なので、注意深い聴診、心電図による右心負荷、心臓超音波検査は重要です。黄疸よりも、心室中隔欠損症やファロー四徴症などの心奇形が前景にたつことがあり、CATCH22症候群との鑑別が問題になることもあります。心臓外の血管障害(脳動脈瘤・もやもや病・腎血管狭窄など)が見られ、頭蓋内出血に至る症例も数多く報告されているので、MRアンギオグラフィーや心臓カテーテル検査の際の脳血管検索も有用です。
(3)顔貌の変化:9割以上に広い前額、幅広い鼻梁、彫りの深い眼、小さなオトガイなどが見られます。(写真1)
(4)骨格系:約半数でButterfly vertebraが見られます。閉塞性黄疸の乳児では、直ぐに骨に合わせて椎体のX線撮影を行いましょう。
(5)眼病変:約8割に後部胎生環が見られ診断に有用なので、AGSを疑う場合は眼科検診を行います。しかし、後部胎生環はAGS以外でもしばしば認められる所見です。
(6)腎の異常:尿細管アシドーシス、腎機能低下などが合併することがあります。AGS2では腎障害が全例に見られ、重症なことが特徴的とされています。
(7)このほか、発達障害、思春期遅延、膵機能不全、難聴など多彩な症状が報告されています。
◆遺伝子変異から臨床的な重症度を推測することは不可能なので、出生前診断に遺伝子診断を用いることは困難です。
<治療と予後>
症例ごとに臓器障害の重症度に大きな違いがあります。例えば、同じ遺伝子変異を有する同一家系内のメンバーに、無症状例と肝移植の必要な患者が併存することもあります。したがって治療方針を立てるためには、各臓器について重症度を把握することが大切です。胆汁うっ滞、心血管奇形、腎機能低下やその他の臓器障害が治療の対象となります。
肝移植について(図6、7):黄疸を伴うAGS患児の約3分の1で肝移植が必要になると言われています。1歳を過ぎても高度の顕性黄疸や高度の高コレステロール血症が持続する症例で肝移植の適応になりやすいとされているので、このような場合は肝移植施設と相談すると良いでしょう。成長障害、強い皮膚掻痒感、高度の黄色腫、病的骨折、肝不全徴候が肝移植の適応となります。適応を決める前に、心血管奇形や腎障害の重症度を評価します。両親のどちらかが生体肝移植のドナーになることを希望した場合は、軽症のAGSでないか精査が必要です。肝生検やMRCPによる肝外胆管の評価、遺伝子検査などが行われる場合があります。肝移植が成功すれば皮膚掻痒、高脂血症、黄色腫は改善します。末梢性肺動脈狭窄が高度の場合は、肝移植後にも狭窄の進行に注意が必要です。短期的には成長障害も改善すると期待されますが、長期的にはAGSによって成長障害をきたす場合が多いです。
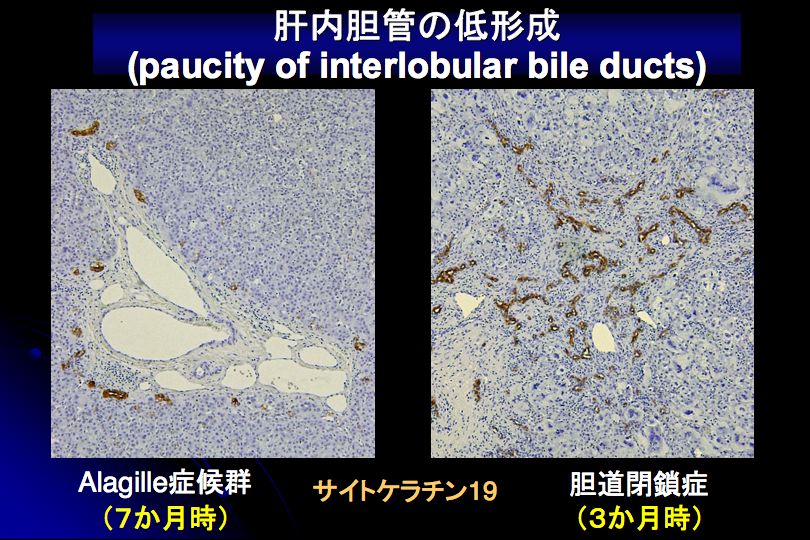 [図6] ※クリックで拡大できます |
 [図7] ※クリックで拡大できます |
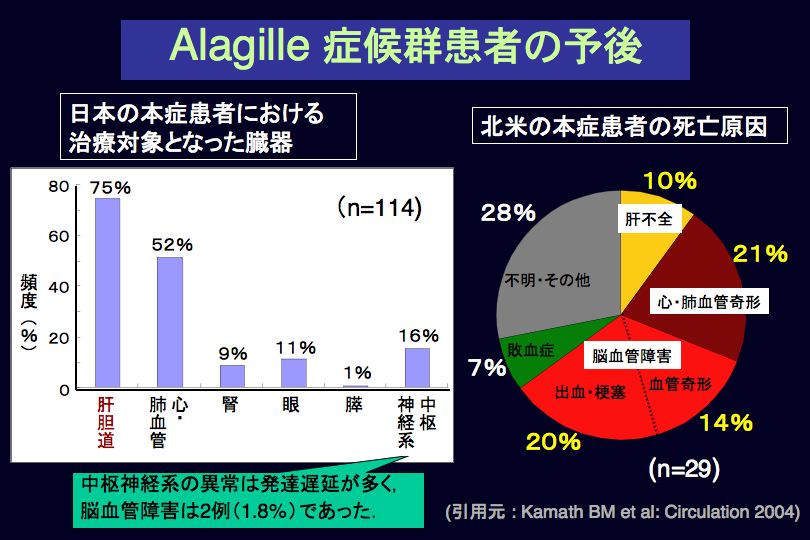
[図8] ※クリックで拡大できます
予後について:日本におけるAGS患者さんの抱える問題点と米国における死亡原因を図8に示します。
Copyright©2011 Alagille syndrome and infantile cholestasis in Japan. All Rights Reserved.