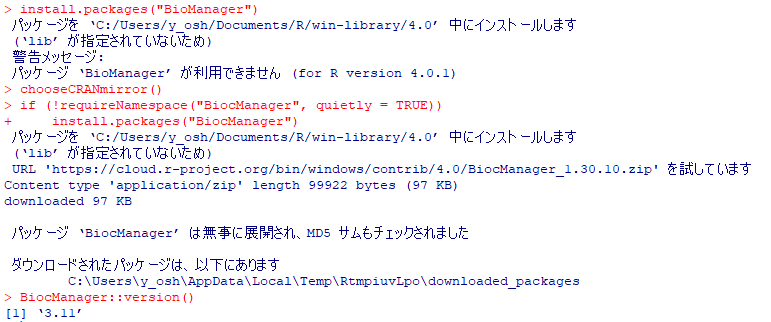
Bioconductorをアップデートしようとしたのですが、現時点の最新バージョン3.11は、Rのバージョンがちょこっと古くてインストールできません。そこで、3.10を入れることにしました。以下はその時のログで、エラーではじかれるところも含めて掲載です。(winのディレクトリ名だけ変更しています)
[Workspace loaded from ~/.RData]
> library(BiocManager)
Bioconductor version 3.9 (BiocManager 1.30.10), ?BiocManager::install for help
Bioconductor version ‘3.9’ is out-of-date; the current release version ‘3.11’
is available with R version ‘4.0’; see https://bioconductor.org/install
警告メッセージ:
パッケージ ‘BiocManager’ はバージョン 3.6.3 の R の下で造られました
> BiocManager::install(version = “3.11”)
エラー: Bioconductor version ‘3.11’ requires R version ‘4.0’; see
https://bioconductor.org/install
> BiocManager::install(version = “3.10”)
Upgrade 195 packages to Bioconductor version ‘3.10’? [y/n]:
y
Bioconductor version 3.10 (BiocManager 1.30.10), R 3.6.1 (2019-07-05)
Installing package(s) ‘BiocVersion’
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/BiocVersion_3.10.1.zip’ を試しています
Content type ‘application/zip’ length 8903 bytes
downloaded 8903 bytes
package ‘BiocVersion’ successfully unpacked and MD5 sums checked
unpackPkgZip(foundpkgs[okp, 2L], foundpkgs[okp, 1L], lib, libs_only, でエラー:
ERROR: failed to lock directory ‘C:\Users\my_username\Documents\R\win-library\3.6’ for modifying
Try removing ‘C:\Users\my_username\Documents\R\win-library\3.6/00LOCK’
追加情報: 警告メッセージ:
1: readChar(…) で:
UTF-8 以外のマルチバイトロケールではバイト型でのみ読み込み可能です
2: readChar(…) で:
UTF-8 以外のマルチバイトロケールではバイト型でのみ読み込み可能です
3: readChar(…) で:
UTF-8 以外のマルチバイトロケールではバイト型でのみ読み込み可能です
4: readChar(…) で:
UTF-8 以外のマルチバイトロケールではバイト型でのみ読み込み可能です
5: readChar(…) で:
UTF-8 以外のマルチバイトロケールではバイト型でのみ読み込み可能です
> BiocManager::install(version = “3.10”)
Upgrade 195 packages to Bioconductor version ‘3.10’? [y/n]:
y
Bioconductor version 3.10 (BiocManager 1.30.10), R 3.6.1 (2019-07-05)
Installing package(s) ‘BiocVersion’
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/BiocVersion_3.10.1.zip’ を試しています
Content type ‘application/zip’ length 8903 bytes
downloaded 8903 bytes
package ‘BiocVersion’ successfully unpacked and MD5 sums checked
The downloaded binary packages are in
C:\Users\my_username\AppData\Local\Temp\RtmpQhgRhw\downloaded_packages
Installing package(s) ‘wordcloud2’, ‘annotate’, ‘AnnotationDbi’, ‘backports’,
‘BH’, ‘Biobase’, ‘BiocGenerics’, ‘BiocParallel’, ‘BiocVersion’, ‘bit’,
‘blob’, ‘broom’, ‘Cairo’, ‘callr’, ‘car’, ‘carData’, ‘caTools’, ‘checkmate’,
‘classInt’, ‘cli’, ‘covr’, ‘crosstalk’, ‘curl’, ‘data.table’, ‘DBI’,
‘dbplyr’, ‘DelayedArray’, ‘DESeq2’, ‘devtools’, ‘DiagrammeR’, ‘digest’,
‘dplyr’, ‘drat’, ‘DT’, ‘edgeR’, ‘ellipsis’, ‘epiR’, ‘fansi’, ‘farver’,
‘fitdistrplus’, ‘forcats’, ‘foreach’, ‘forecast’, ‘fracdiff’, ‘fs’, ‘gargle’,
‘genefilter’, ‘geneplotter’, ‘GenomeInfoDb’, ‘GenomeInfoDbData’,
‘GenomicRanges’, ‘GEOmetadb’, ‘GEOquery’, ‘ggplot2’, ‘ggpubr’, ‘ggrepel’,
‘gh’, ‘git2r’, ‘glue’, ‘GO.db’, ‘googleAuthR’, ‘googledrive’,
‘googlesheets4’, ‘gplots’, ‘gtools’, ‘h2o’, ‘haven’, ‘Heatplus’, ‘hexbin’,
‘Hmisc’, ‘hms’, ‘htmlTable’, ‘htmltools’, ‘httpuv’, ‘igraph’, ‘imager’,
‘IRanges’, ‘ISOcodes’, ‘jomo’, ‘jsonlite’, ‘knitr’, ‘labelled’, ‘lamW’,
‘later’, ‘latticeExtra’, ‘LBE’, ‘lifecycle’, ‘limma’, ‘lme4’, ‘lmerTest’,
‘locfit’, ‘lubridate’, ‘maptools’, ‘matrixStats’, ‘mcmc’, ‘MCMCpack’,
‘MetaIntegrator’, ‘mice’, ‘mime’, ‘modelr’, ‘modeltools’, ‘multtest’,
‘mvtnorm’, ‘nloptr’, ‘openxlsx’, ‘ordinal’, ‘org.Hs.eg.db’, ‘pbkrtest’,
‘pillar’, ‘pkgbuild’, ‘pkgload’, ‘plotly’, ‘plyr’, ‘pracma’,
‘preprocessCore’, ‘prettyunits’, ‘pROC’, ‘processx’, ‘promises’, ‘ps’,
‘purrr’, ‘quantmod’, ‘quantreg’, ‘R.methodsS3’, ‘raster’, ‘Rcpp’,
‘RcppArmadillo’, ‘RcppEigen’, ‘RcppParallel’, ‘RcppProgress’, ‘RCurl’,
‘rematch2’, ‘remotes’, ‘reshape2’, ‘rex’, ‘rgdal’, ‘rgexf’, ‘rgl’, ‘rlang’,
‘rmarkdown’, ‘RMySQL’, ‘ROCit’, ‘ROCR’, ‘roxygen2’, ‘RSpectra’, ‘RSQLite’,
‘rstudioapi’, ‘rversions’, ‘S4Vectors’, ‘scales’, ‘selectr’, ‘seqminer’,
‘SeqNet’, ‘sf’, ‘shiny’, ‘SKAT’, ‘slam’, ‘sn’, ‘sp’, ‘SparseM’, ‘spatstat’,
‘spData’, ‘statmod’, ‘stopwords’, ‘stringi’, ‘SummarizedExperiment’,
‘surveillance’, ‘survey’, ‘tableone’, ‘testthat’, ‘text2vec’, ‘tibble’,
‘tidyr’, ‘tidyselect’, ‘tidyverse’, ‘tinytex’, ‘tm’, ‘topicmodels’, ‘TTR’,
‘units’, ‘usethis’, ‘uuid’, ‘vctrs’, ‘visNetwork’, ‘withr’, ‘xfun’,
‘xgboost’, ‘XML’, ‘xml2’, ‘xts’, ‘XVector’, ‘yaml’, ‘zlibbioc’, ‘zoo’
There are binary versions available but the source versions are later:
binary source needs_compilation
backports 1.1.7 1.1.8 TRUE
htmlTable 1.13.3 2.0.0 FALSE
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/wordcloud2_0.2.1.zip’ を試しています
Content type ‘application/zip’ length 999844 bytes (976 KB)
downloaded 976 KB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/annotate_1.64.0.zip’ を試しています
Content type ‘application/zip’ length 2238722 bytes (2.1 MB)
downloaded 2.1 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/AnnotationDbi_1.48.0.zip’ を試しています
Content type ‘application/zip’ length 5226197 bytes (5.0 MB)
downloaded 5.0 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/BH_1.72.0-3.zip’ を試しています
Content type ‘application/zip’ length 18270741 bytes (17.4 MB)
downloaded 17.4 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/Biobase_2.46.0.zip’ を試しています
Content type ‘application/zip’ length 2421436 bytes (2.3 MB)
downloaded 2.3 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/BiocGenerics_0.32.0.zip’ を試しています
Content type ‘application/zip’ length 748769 bytes (731 KB)
downloaded 731 KB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/BiocParallel_1.20.1.zip’ を試しています
Content type ‘application/zip’ length 1672038 bytes (1.6 MB)
downloaded 1.6 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/BiocVersion_3.10.1.zip’ を試しています
Content type ‘application/zip’ length 8903 bytes
downloaded 8903 bytes
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/bit_1.1-15.2.zip’ を試しています
Content type ‘application/zip’ length 252475 bytes (246 KB)
downloaded 246 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/blob_1.2.1.zip’ を試しています
Content type ‘application/zip’ length 47723 bytes (46 KB)
downloaded 46 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/broom_0.5.6.zip’ を試しています
Content type ‘application/zip’ length 1970639 bytes (1.9 MB)
downloaded 1.9 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/Cairo_1.5-12.zip’ を試しています
Content type ‘application/zip’ length 1040911 bytes (1016 KB)
downloaded 1016 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/callr_3.4.3.zip’ を試しています
Content type ‘application/zip’ length 390402 bytes (381 KB)
downloaded 381 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/car_3.0-8.zip’ を試しています
Content type ‘application/zip’ length 1564099 bytes (1.5 MB)
downloaded 1.5 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/carData_3.0-4.zip’ を試しています
Content type ‘application/zip’ length 1821316 bytes (1.7 MB)
downloaded 1.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/caTools_1.18.0.zip’ を試しています
Content type ‘application/zip’ length 330432 bytes (322 KB)
downloaded 322 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/checkmate_2.0.0.zip’ を試しています
Content type ‘application/zip’ length 704639 bytes (688 KB)
downloaded 688 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/classInt_0.4-3.zip’ を試しています
Content type ‘application/zip’ length 467957 bytes (456 KB)
downloaded 456 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/cli_2.0.2.zip’ を試しています
Content type ‘application/zip’ length 397888 bytes (388 KB)
downloaded 388 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/covr_3.5.0.zip’ を試しています
Content type ‘application/zip’ length 305301 bytes (298 KB)
downloaded 298 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/crosstalk_1.1.0.1.zip’ を試しています
Content type ‘application/zip’ length 778716 bytes (760 KB)
downloaded 760 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/curl_4.3.zip’ を試しています
Content type ‘application/zip’ length 4127987 bytes (3.9 MB)
downloaded 3.9 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/data.table_1.12.8.zip’ を試しています
Content type ‘application/zip’ length 2276767 bytes (2.2 MB)
downloaded 2.2 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/DBI_1.1.0.zip’ を試しています
Content type ‘application/zip’ length 607347 bytes (593 KB)
downloaded 593 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/dbplyr_1.4.4.zip’ を試しています
Content type ‘application/zip’ length 644261 bytes (629 KB)
downloaded 629 KB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/DelayedArray_0.12.3.zip’ を試しています
Content type ‘application/zip’ length 2261536 bytes (2.2 MB)
downloaded 2.2 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/DESeq2_1.26.0.zip’ を試しています
Content type ‘application/zip’ length 2552125 bytes (2.4 MB)
downloaded 2.4 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/devtools_2.3.0.zip’ を試しています
Content type ‘application/zip’ length 350962 bytes (342 KB)
downloaded 342 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/DiagrammeR_1.0.6.1.zip’ を試しています
Content type ‘application/zip’ length 5558172 bytes (5.3 MB)
downloaded 5.3 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/digest_0.6.25.zip’ を試しています
Content type ‘application/zip’ length 249475 bytes (243 KB)
downloaded 243 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/dplyr_1.0.0.zip’ を試しています
Content type ‘application/zip’ length 1522184 bytes (1.5 MB)
downloaded 1.5 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/drat_0.1.6.zip’ を試しています
Content type ‘application/zip’ length 89567 bytes (87 KB)
downloaded 87 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/DT_0.13.zip’ を試しています
Content type ‘application/zip’ length 1734181 bytes (1.7 MB)
downloaded 1.7 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/edgeR_3.28.1.zip’ を試しています
Content type ‘application/zip’ length 2986014 bytes (2.8 MB)
downloaded 2.8 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/ellipsis_0.3.1.zip’ を試しています
Content type ‘application/zip’ length 45934 bytes (44 KB)
downloaded 44 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/epiR_1.0-15.zip’ を試しています
Content type ‘application/zip’ length 857628 bytes (837 KB)
downloaded 837 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/fansi_0.4.1.zip’ を試しています
Content type ‘application/zip’ length 223884 bytes (218 KB)
downloaded 218 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/farver_2.0.3.zip’ を試しています
Content type ‘application/zip’ length 1912126 bytes (1.8 MB)
downloaded 1.8 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/fitdistrplus_1.1-1.zip’ を試しています
Content type ‘application/zip’ length 2725046 bytes (2.6 MB)
downloaded 2.6 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/forcats_0.5.0.zip’ を試しています
Content type ‘application/zip’ length 356323 bytes (347 KB)
downloaded 347 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/foreach_1.5.0.zip’ を試しています
Content type ‘application/zip’ length 145920 bytes (142 KB)
downloaded 142 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/forecast_8.12.zip’ を試しています
Content type ‘application/zip’ length 2358698 bytes (2.2 MB)
downloaded 2.2 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/fracdiff_1.5-1.zip’ を試しています
Content type ‘application/zip’ length 135022 bytes (131 KB)
downloaded 131 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/fs_1.4.1.zip’ を試しています
Content type ‘application/zip’ length 773609 bytes (755 KB)
downloaded 755 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/gargle_0.5.0.zip’ を試しています
Content type ‘application/zip’ length 400070 bytes (390 KB)
downloaded 390 KB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/genefilter_1.68.0.zip’ を試しています
Content type ‘application/zip’ length 1540166 bytes (1.5 MB)
downloaded 1.5 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/geneplotter_1.64.0.zip’ を試しています
Content type ‘application/zip’ length 1585192 bytes (1.5 MB)
downloaded 1.5 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/GenomeInfoDb_1.22.1.zip’ を試しています
Content type ‘application/zip’ length 3839163 bytes (3.7 MB)
downloaded 3.7 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/GenomicRanges_1.38.0.zip’ を試しています
Content type ‘application/zip’ length 2198139 bytes (2.1 MB)
downloaded 2.1 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/GEOquery_2.54.1.zip’ を試しています
Content type ‘application/zip’ length 13850910 bytes (13.2 MB)
downloaded 13.2 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/ggplot2_3.3.2.zip’ を試しています
Content type ‘application/zip’ length 4068432 bytes (3.9 MB)
downloaded 3.9 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/ggpubr_0.3.0.zip’ を試しています
Content type ‘application/zip’ length 1859849 bytes (1.8 MB)
downloaded 1.8 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/ggrepel_0.8.2.zip’ を試しています
Content type ‘application/zip’ length 1154089 bytes (1.1 MB)
downloaded 1.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/gh_1.1.0.zip’ を試しています
Content type ‘application/zip’ length 110365 bytes (107 KB)
downloaded 107 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/git2r_0.27.1.zip’ を試しています
Content type ‘application/zip’ length 3687960 bytes (3.5 MB)
downloaded 3.5 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/glue_1.4.1.zip’ を試しています
Content type ‘application/zip’ length 154028 bytes (150 KB)
downloaded 150 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/googleAuthR_1.3.0.zip’ を試しています
Content type ‘application/zip’ length 406479 bytes (396 KB)
downloaded 396 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/googledrive_1.0.1.zip’ を試しています
Content type ‘application/zip’ length 1770548 bytes (1.7 MB)
downloaded 1.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/googlesheets4_0.2.0.zip’ を試しています
Content type ‘application/zip’ length 467471 bytes (456 KB)
downloaded 456 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/gplots_3.0.3.zip’ を試しています
Content type ‘application/zip’ length 595339 bytes (581 KB)
downloaded 581 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/gtools_3.8.2.zip’ を試しています
Content type ‘application/zip’ length 336453 bytes (328 KB)
downloaded 328 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/h2o_3.30.0.1.zip’ を試しています
Content type ‘application/zip’ length 130944314 bytes (124.9 MB)
downloaded 124.9 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/haven_2.3.1.zip’ を試しています
Content type ‘application/zip’ length 1067204 bytes (1.0 MB)
downloaded 1.0 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/Heatplus_2.32.1.zip’ を試しています
Content type ‘application/zip’ length 1007868 bytes (984 KB)
downloaded 984 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/hexbin_1.28.1.zip’ を試しています
Content type ‘application/zip’ length 1002143 bytes (978 KB)
downloaded 978 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/Hmisc_4.4-0.zip’ を試しています
Content type ‘application/zip’ length 3167446 bytes (3.0 MB)
downloaded 3.0 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/hms_0.5.3.zip’ を試しています
Content type ‘application/zip’ length 109783 bytes (107 KB)
downloaded 107 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/htmltools_0.5.0.zip’ を試しています
Content type ‘application/zip’ length 233079 bytes (227 KB)
downloaded 227 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/httpuv_1.5.4.zip’ を試しています
Content type ‘application/zip’ length 1551610 bytes (1.5 MB)
downloaded 1.5 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/igraph_1.2.5.zip’ を試しています
Content type ‘application/zip’ length 9165651 bytes (8.7 MB)
downloaded 8.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/imager_0.42.3.zip’ を試しています
Content type ‘application/zip’ length 10031218 bytes (9.6 MB)
downloaded 9.6 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/IRanges_2.20.2.zip’ を試しています
Content type ‘application/zip’ length 2405448 bytes (2.3 MB)
downloaded 2.3 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/ISOcodes_2020.03.16.zip’ を試しています
Content type ‘application/zip’ length 306800 bytes (299 KB)
downloaded 299 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/jomo_2.7-1.zip’ を試しています
Content type ‘application/zip’ length 2245621 bytes (2.1 MB)
downloaded 2.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/jsonlite_1.6.1.zip’ を試しています
Content type ‘application/zip’ length 1165739 bytes (1.1 MB)
downloaded 1.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/knitr_1.28.zip’ を試しています
Content type ‘application/zip’ length 1431167 bytes (1.4 MB)
downloaded 1.4 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/labelled_2.5.0.zip’ を試しています
Content type ‘application/zip’ length 210283 bytes (205 KB)
downloaded 205 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/lamW_1.3.2.zip’ を試しています
Content type ‘application/zip’ length 539827 bytes (527 KB)
downloaded 527 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/later_1.1.0.1.zip’ を試しています
Content type ‘application/zip’ length 687561 bytes (671 KB)
downloaded 671 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/latticeExtra_0.6-29.zip’ を試しています
Content type ‘application/zip’ length 2202331 bytes (2.1 MB)
downloaded 2.1 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/LBE_1.54.0.zip’ を試しています
Content type ‘application/zip’ length 453112 bytes (442 KB)
downloaded 442 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/lifecycle_0.2.0.zip’ を試しています
Content type ‘application/zip’ length 101697 bytes (99 KB)
downloaded 99 KB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/limma_3.42.2.zip’ を試しています
Content type ‘application/zip’ length 3096814 bytes (3.0 MB)
downloaded 3.0 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/lme4_1.1-23.zip’ を試しています
Content type ‘application/zip’ length 5708494 bytes (5.4 MB)
downloaded 5.4 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/lmerTest_3.1-2.zip’ を試しています
Content type ‘application/zip’ length 417383 bytes (407 KB)
downloaded 407 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/locfit_1.5-9.4.zip’ を試しています
Content type ‘application/zip’ length 689589 bytes (673 KB)
downloaded 673 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/lubridate_1.7.9.zip’ を試しています
Content type ‘application/zip’ length 1605006 bytes (1.5 MB)
downloaded 1.5 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/maptools_1.0-1.zip’ を試しています
Content type ‘application/zip’ length 2175494 bytes (2.1 MB)
downloaded 2.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/matrixStats_0.56.0.zip’ を試しています
Content type ‘application/zip’ length 1727460 bytes (1.6 MB)
downloaded 1.6 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/mcmc_0.9-7.zip’ を試しています
Content type ‘application/zip’ length 1454314 bytes (1.4 MB)
downloaded 1.4 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/MCMCpack_1.4-8.zip’ を試しています
Content type ‘application/zip’ length 3257596 bytes (3.1 MB)
downloaded 3.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/MetaIntegrator_2.1.3.zip’ を試しています
Content type ‘application/zip’ length 3531328 bytes (3.4 MB)
downloaded 3.4 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/mice_3.9.0.zip’ を試しています
Content type ‘application/zip’ length 1776683 bytes (1.7 MB)
downloaded 1.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/mime_0.9.zip’ を試しています
Content type ‘application/zip’ length 48395 bytes (47 KB)
downloaded 47 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/modelr_0.1.8.zip’ を試しています
Content type ‘application/zip’ length 203039 bytes (198 KB)
downloaded 198 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/modeltools_0.2-23.zip’ を試しています
Content type ‘application/zip’ length 206077 bytes (201 KB)
downloaded 201 KB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/multtest_2.42.0.zip’ を試しています
Content type ‘application/zip’ length 957511 bytes (935 KB)
downloaded 935 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/mvtnorm_1.1-1.zip’ を試しています
Content type ‘application/zip’ length 271534 bytes (265 KB)
downloaded 265 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/nloptr_1.2.2.1.zip’ を試しています
Content type ‘application/zip’ length 1079130 bytes (1.0 MB)
downloaded 1.0 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/openxlsx_4.1.5.zip’ を試しています
Content type ‘application/zip’ length 2601318 bytes (2.5 MB)
downloaded 2.5 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/ordinal_2019.12-10.zip’ を試しています
Content type ‘application/zip’ length 1244142 bytes (1.2 MB)
downloaded 1.2 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/pbkrtest_0.4-8.6.zip’ を試しています
Content type ‘application/zip’ length 275920 bytes (269 KB)
downloaded 269 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/pillar_1.4.4.zip’ を試しています
Content type ‘application/zip’ length 186096 bytes (181 KB)
downloaded 181 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/pkgbuild_1.0.8.zip’ を試しています
Content type ‘application/zip’ length 138864 bytes (135 KB)
downloaded 135 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/pkgload_1.1.0.zip’ を試しています
Content type ‘application/zip’ length 163036 bytes (159 KB)
downloaded 159 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/plotly_4.9.2.1.zip’ を試しています
Content type ‘application/zip’ length 3044630 bytes (2.9 MB)
downloaded 2.9 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/plyr_1.8.6.zip’ を試しています
Content type ‘application/zip’ length 1310998 bytes (1.3 MB)
downloaded 1.3 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/pracma_2.2.9.zip’ を試しています
Content type ‘application/zip’ length 1739254 bytes (1.7 MB)
downloaded 1.7 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/preprocessCore_1.48.0.zip’ を試しています
Content type ‘application/zip’ length 252626 bytes (246 KB)
downloaded 246 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/prettyunits_1.1.1.zip’ を試しています
Content type ‘application/zip’ length 37684 bytes (36 KB)
downloaded 36 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/pROC_1.16.2.zip’ を試しています
Content type ‘application/zip’ length 1326542 bytes (1.3 MB)
downloaded 1.3 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/processx_3.4.2.zip’ を試しています
Content type ‘application/zip’ length 428127 bytes (418 KB)
downloaded 418 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/promises_1.1.1.zip’ を試しています
Content type ‘application/zip’ length 1787064 bytes (1.7 MB)
downloaded 1.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/ps_1.3.3.zip’ を試しています
Content type ‘application/zip’ length 333411 bytes (325 KB)
downloaded 325 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/purrr_0.3.4.zip’ を試しています
Content type ‘application/zip’ length 430973 bytes (420 KB)
downloaded 420 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/quantmod_0.4.17.zip’ を試しています
Content type ‘application/zip’ length 965229 bytes (942 KB)
downloaded 942 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/quantreg_5.55.zip’ を試しています
Content type ‘application/zip’ length 1782051 bytes (1.7 MB)
downloaded 1.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/R.methodsS3_1.8.0.zip’ を試しています
Content type ‘application/zip’ length 81353 bytes (79 KB)
downloaded 79 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/raster_3.1-5.zip’ を試しています
Content type ‘application/zip’ length 3344533 bytes (3.2 MB)
downloaded 3.2 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/Rcpp_1.0.4.6.zip’ を試しています
Content type ‘application/zip’ length 3030954 bytes (2.9 MB)
downloaded 2.9 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/RcppArmadillo_0.9.900.1.0.zip’ を試しています
Content type ‘application/zip’ length 2377181 bytes (2.3 MB)
downloaded 2.3 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/RcppEigen_0.3.3.7.0.zip’ を試しています
Content type ‘application/zip’ length 2678957 bytes (2.6 MB)
downloaded 2.6 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/RcppParallel_5.0.1.zip’ を試しています
Content type ‘application/zip’ length 3757488 bytes (3.6 MB)
downloaded 3.6 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/RcppProgress_0.4.2.zip’ を試しています
Content type ‘application/zip’ length 33942 bytes (33 KB)
downloaded 33 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/RCurl_1.98-1.2.zip’ を試しています
Content type ‘application/zip’ length 2987986 bytes (2.8 MB)
downloaded 2.8 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/rematch2_2.1.2.zip’ を試しています
Content type ‘application/zip’ length 47552 bytes (46 KB)
downloaded 46 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/remotes_2.1.1.zip’ を試しています
Content type ‘application/zip’ length 358530 bytes (350 KB)
downloaded 350 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/reshape2_1.4.4.zip’ を試しています
Content type ‘application/zip’ length 630986 bytes (616 KB)
downloaded 616 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/rex_1.2.0.zip’ を試しています
Content type ‘application/zip’ length 125491 bytes (122 KB)
downloaded 122 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/rgdal_1.5-10.zip’ を試しています
Content type ‘application/zip’ length 36493604 bytes (34.8 MB)
downloaded 34.8 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/rgexf_0.16.0.zip’ を試しています
Content type ‘application/zip’ length 267948 bytes (261 KB)
downloaded 261 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/rgl_0.100.54.zip’ を試しています
Content type ‘application/zip’ length 4306027 bytes (4.1 MB)
downloaded 4.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/rlang_0.4.6.zip’ を試しています
Content type ‘application/zip’ length 1128385 bytes (1.1 MB)
downloaded 1.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/rmarkdown_2.3.zip’ を試しています
Content type ‘application/zip’ length 3632281 bytes (3.5 MB)
downloaded 3.5 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/RMySQL_0.10.20.zip’ を試しています
Content type ‘application/zip’ length 3206152 bytes (3.1 MB)
downloaded 3.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/ROCit_2.1.1.zip’ を試しています
Content type ‘application/zip’ length 291305 bytes (284 KB)
downloaded 284 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/ROCR_1.0-11.zip’ を試しています
Content type ‘application/zip’ length 405720 bytes (396 KB)
downloaded 396 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/roxygen2_7.1.0.zip’ を試しています
Content type ‘application/zip’ length 1178727 bytes (1.1 MB)
downloaded 1.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/RSpectra_0.16-0.zip’ を試しています
Content type ‘application/zip’ length 1278659 bytes (1.2 MB)
downloaded 1.2 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/RSQLite_2.2.0.zip’ を試しています
Content type ‘application/zip’ length 2273874 bytes (2.2 MB)
downloaded 2.2 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/rstudioapi_0.11.zip’ を試しています
Content type ‘application/zip’ length 280393 bytes (273 KB)
downloaded 273 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/rversions_2.0.2.zip’ を試しています
Content type ‘application/zip’ length 68785 bytes (67 KB)
downloaded 67 KB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/S4Vectors_0.24.4.zip’ を試しています
Content type ‘application/zip’ length 2140759 bytes (2.0 MB)
downloaded 2.0 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/scales_1.1.1.zip’ を試しています
Content type ‘application/zip’ length 561984 bytes (548 KB)
downloaded 548 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/selectr_0.4-2.zip’ を試しています
Content type ‘application/zip’ length 494437 bytes (482 KB)
downloaded 482 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/seqminer_8.0.zip’ を試しています
Content type ‘application/zip’ length 4227867 bytes (4.0 MB)
downloaded 4.0 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/SeqNet_1.1.0.zip’ を試しています
Content type ‘application/zip’ length 7963516 bytes (7.6 MB)
downloaded 7.6 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/sf_0.9-4.zip’ を試しています
Content type ‘application/zip’ length 42635208 bytes (40.7 MB)
downloaded 40.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/shiny_1.4.0.2.zip’ を試しています
Content type ‘application/zip’ length 4942535 bytes (4.7 MB)
downloaded 4.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/SKAT_2.0.0.zip’ を試しています
Content type ‘application/zip’ length 1716196 bytes (1.6 MB)
downloaded 1.6 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/slam_0.1-47.zip’ を試しています
Content type ‘application/zip’ length 211121 bytes (206 KB)
downloaded 206 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/sn_1.6-2.zip’ を試しています
Content type ‘application/zip’ length 1541092 bytes (1.5 MB)
downloaded 1.5 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/sp_1.4-2.zip’ を試しています
Content type ‘application/zip’ length 1891448 bytes (1.8 MB)
downloaded 1.8 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/SparseM_1.78.zip’ を試しています
Content type ‘application/zip’ length 1070197 bytes (1.0 MB)
downloaded 1.0 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/spatstat_1.64-1.zip’ を試しています
Content type ‘application/zip’ length 16947503 bytes (16.2 MB)
downloaded 16.2 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/spData_0.3.5.zip’ を試しています
Content type ‘application/zip’ length 3688560 bytes (3.5 MB)
downloaded 3.5 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/statmod_1.4.34.zip’ を試しています
Content type ‘application/zip’ length 285330 bytes (278 KB)
downloaded 278 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/stopwords_2.0.zip’ を試しています
Content type ‘application/zip’ length 191783 bytes (187 KB)
downloaded 187 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/stringi_1.4.6.zip’ を試しています
Content type ‘application/zip’ length 15310634 bytes (14.6 MB)
downloaded 14.6 MB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/SummarizedExperiment_1.16.1.zip’ を試しています
Content type ‘application/zip’ length 2834911 bytes (2.7 MB)
downloaded 2.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/surveillance_1.18.0.zip’ を試しています
Content type ‘application/zip’ length 6275336 bytes (6.0 MB)
downloaded 6.0 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/survey_4.0.zip’ を試しています
Content type ‘application/zip’ length 2530626 bytes (2.4 MB)
downloaded 2.4 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/tableone_0.11.1.zip’ を試しています
Content type ‘application/zip’ length 254139 bytes (248 KB)
downloaded 248 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/testthat_2.3.2.zip’ を試しています
Content type ‘application/zip’ length 1799253 bytes (1.7 MB)
downloaded 1.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/text2vec_0.6.zip’ を試しています
Content type ‘application/zip’ length 5102940 bytes (4.9 MB)
downloaded 4.9 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/tibble_3.0.1.zip’ を試しています
Content type ‘application/zip’ length 411975 bytes (402 KB)
downloaded 402 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/tidyr_1.1.0.zip’ を試しています
Content type ‘application/zip’ length 1334023 bytes (1.3 MB)
downloaded 1.3 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/tidyselect_1.1.0.zip’ を試しています
Content type ‘application/zip’ length 203224 bytes (198 KB)
downloaded 198 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/tidyverse_1.3.0.zip’ を試しています
Content type ‘application/zip’ length 440174 bytes (429 KB)
downloaded 429 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/tinytex_0.24.zip’ を試しています
Content type ‘application/zip’ length 109882 bytes (107 KB)
downloaded 107 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/tm_0.7-7.zip’ を試しています
Content type ‘application/zip’ length 1372416 bytes (1.3 MB)
downloaded 1.3 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/topicmodels_0.2-11.zip’ を試しています
Content type ‘application/zip’ length 1562988 bytes (1.5 MB)
downloaded 1.5 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/TTR_0.23-6.zip’ を試しています
Content type ‘application/zip’ length 521426 bytes (509 KB)
downloaded 509 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/units_0.6-7.zip’ を試しています
Content type ‘application/zip’ length 1758397 bytes (1.7 MB)
downloaded 1.7 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/usethis_1.6.1.zip’ を試しています
Content type ‘application/zip’ length 603163 bytes (589 KB)
downloaded 589 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/uuid_0.1-4.zip’ を試しています
Content type ‘application/zip’ length 34014 bytes (33 KB)
downloaded 33 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/vctrs_0.3.1.zip’ を試しています
Content type ‘application/zip’ length 1182692 bytes (1.1 MB)
downloaded 1.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/visNetwork_2.0.9.zip’ を試しています
Content type ‘application/zip’ length 4595214 bytes (4.4 MB)
downloaded 4.4 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/withr_2.2.0.zip’ を試しています
Content type ‘application/zip’ length 206490 bytes (201 KB)
downloaded 201 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/xfun_0.15.zip’ を試しています
Content type ‘application/zip’ length 229245 bytes (223 KB)
downloaded 223 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/xgboost_1.1.1.1.zip’ を試しています
Content type ‘application/zip’ length 2743647 bytes (2.6 MB)
downloaded 2.6 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/XML_3.99-0.3.zip’ を試しています
Content type ‘application/zip’ length 4255589 bytes (4.1 MB)
downloaded 4.1 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/xml2_1.3.2.zip’ を試しています
Content type ‘application/zip’ length 3177206 bytes (3.0 MB)
downloaded 3.0 MB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/xts_0.12-0.zip’ を試しています
Content type ‘application/zip’ length 966258 bytes (943 KB)
downloaded 943 KB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/XVector_0.26.0.zip’ を試しています
Content type ‘application/zip’ length 714483 bytes (697 KB)
downloaded 697 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/yaml_2.2.1.zip’ を試しています
Content type ‘application/zip’ length 206516 bytes (201 KB)
downloaded 201 KB
URL ‘https://bioconductor.org/packages/3.10/bioc/bin/windows/contrib/3.6/zlibbioc_1.32.0.zip’ を試しています
Content type ‘application/zip’ length 374998 bytes (366 KB)
downloaded 366 KB
URL ‘https://cran.rstudio.com/bin/windows/contrib/3.6/zoo_1.8-8.zip’ を試しています
Content type ‘application/zip’ length 1096019 bytes (1.0 MB)
downloaded 1.0 MB
package ‘wordcloud2’ successfully unpacked and MD5 sums checked
package ‘annotate’ successfully unpacked and MD5 sums checked
package ‘AnnotationDbi’ successfully unpacked and MD5 sums checked
package ‘BH’ successfully unpacked and MD5 sums checked