
<この記事は内科学会雑誌に掲載した記事のセルフアーカイブです。誤字脱字等も含め内容は公開版の最終稿と同一です。>
Evidence-based Medicine (EBM)
有害事象と医薬品の因果関係評価について
大島康雄
東京大学医科学研究所 先端医療研究センター・分子療法分野
郵便番号 108-8639
住所 東京都港区白金台4丁目6-1
℡ 03-6301-3845
電子メール 0-oshima@umin.ac.jp
はじめに
医薬品医療機器総合機構(PMDA)のWebpageによると、2010年1月から11月までの期間に使用上の注意の改訂指示があった医薬品情報は170件あった (http://www.info.pmda.go.jp/kaitei/kaitei_index.html)。 この中でアンジオテンシンII受容体拮抗薬、いわゆるARBである、オルメサルタン メドキソミル、テルミサルタン、バルサルタンおよびそれらを薬効成分として含む合剤の使用上の注意へ、「横紋筋融解症」の副作用の記載をするようにとの指示が私の目を引いた。横紋筋融解症は一旦発現すると死亡に至る割合が1割近くもある深刻な病態である(1)。それに加え、ARBは臨床現場で多くの患者さんへ使用されているため、規制当局からの情報は大きな影響力を有しかねない。しかし、気になったのはそのためだけではない。HMG-CoA還元酵素阻害薬、いわゆるスタチン類では個人的な臨床経験としてあるいは同僚医師らとのコミュニケーションの中で、クレアチンキナーゼ上昇あるいはこれに筋症状等の伴った筋炎と考えられる症例を経験することがあり、スタチンの筋組織に対する傷害性を感じる機会があった。これに対して、ARBではこうした身近な経験をする機会が乏しかったのだ。厚生労働省のWebpage掲載文書である医薬品・医療機器等安全性情報No.271の記載によると、オルメサルタン メドキソミルでは、平成21年度1年間での使用者数おおよそ180万人に対し、集計当時の直近3年間に横紋筋融解症症例のうち因果関係が否定できない症例が1例報告されている、との記載がある。 同じくテルミサルタンでは1年間使用者数190万人に対し直近3年間で3症例、バルサルタンでも同じく410万人に対し4症例との記載である (http://www1.mhlw.go.jp/kinkyu/iyaku_j/iyaku_j/anzenseijyouhou/271-2.pdf)。医薬品・医療機器等安全性情報No.271には改訂指示の根拠となった症例の概要等に関する情報が紹介されている。それぞれの症例の臨床経過を見ると、症例で副作用が報告されていることは理解できるものの、添付文書上で注意喚起を行うという一種のリスクコミュニケーションが必要であると判断したロジックは読み取れない。医薬品・医療機器等安全性情報No.271に記載された数字には、副作用症例を経験された臨床現場の先生方が任意でご報告される、いわゆる自発報告に基づく数字が含まれると考えられる。自発報告の弱点として、発現していても報告されない症例が少なからずあると思われる、いわゆるアンダーレポーティングの問題が指摘されている。言い換えるならば、実際には報告されている症例より多くの有害事象が発現している可能性が考えられる。それにしてもこの程度の報告数であれば身近な経験が情報共有されないのも納得ができる。と同時に、添付文書上で注意喚起を行うというリスクコミュニケーションが本当に必要なのか、情報の受け手としての医師はこの情報をどの様に日々の臨床に生かすことが求められているのか疑問が湧く。
本稿では、医薬品使用中または投与後におきた医療上の好ましくない事象である有害事象が、本当に医薬品が原因で起きた副作用であるかどうか、すなわち有害事象と医薬品の因果関係についてどのような判断の方法があるのかについて、いくつかの例を紹介させていただく。
個別症例の有害事象に関する因果関係
臨床試験中に生じた有害事象と試験薬との因果関係の評価は、重要な意味を持ちうるにもかかわらず、客観的で確立された基準があるとは言えない。疾患の経過中に起きた有害事象は、被疑薬による副作用のほか、被験者がもともと有していた病態に関連した症状、併用薬剤による事象、治療手技の合併症、それらとは無関係に偶然おこる偶発症などさまざまな可能性がある。原因についてのさまざまな可能性を臨床的に推論してゆく中で、相対的に他の要因が考えにくい場合に被疑薬との因果関係があると考えられる。つまり、いわゆる臨床推論そのものであり、一律に基準を設けるのは困難である。治験中に得られる安全性情報の取り扱いについて、INTERNATIONAL CONFERENCE ON HARMONISATION (ICH) E2Aという国際的ガイドラインがある。その規定によると、薬物投与後に起きた有害事象について、完全に否定することは論理的には困難であるにもかかわらず、「因果関係が否定できない場合に、合理性を以て因果関係の可能性があるとする」との考え方が記載されている(2)。脚注 このように基準となるべき文言についても客観的で一定の判断が常にできる基準とは言い難い。このほかにも考慮するべき点はいくつか報告されている。上記ICH E2Aの記述および、同じく国際的な治験や市販後の医薬品評価にかかわる議論を深めてきた国際医科学団体協議会(CIOMS, council for international organizations of medical science)が、因果関係の判断に関して考慮するべきとして指摘している点を4点以下に列挙する。あるべき姿として、評価できるだけの十分な医療上の情報を得たうえで判断がなされるべきである。
- については、被疑薬を再投与することは通常推奨されておらず、情報が得られにくいと思われる。しかし、再投与により再現性が確認できる場合には被疑薬によって有害事象が起きていたとの因果関係が強く支持される。
- 深刻な有害事象が起こり、被疑薬の中止によって有害事象が軽快することが期待される場合には被疑薬が中止される場合が多いであろう。被疑薬の中止によって有害事象が軽快することが通常期待できない発がん等の例を除いて、被偽薬中止によって有害事象が軽快しない場合は、因果関係は考えにくいと判断するかもしれない。
- 好発時期が知られている有害事象、例えばアナフィラキシーショックや抗がん薬の骨髄抑制性の副作用等については、個別症例の発現時期と知られている好発時期とで矛盾がないかが、因果関係を評価するために考慮されるだろう。変異原性試験やがん原生試験等の結果に特に問題となる所見がない薬物については、被疑薬投与開始から1-2年目程度までに発症した悪性疾患については薬剤との因果関係が否定的と考えるだろう。放射線被ばくは遺伝子に障害をもたらすと考えられるが、その放射線被ばくをもたらす原子爆弾投下後の甲状腺がんや白血病の発症のピークが数年後にあるとの調査結果が知られている。また、腫瘤の成長速度に関する基礎的な研究の結果等もふまえ、放射線や薬物への曝露によるイニシエーションから臨床的な発がんには一般的に数年が必要と考えられているからである。
- 報告された有害事象が、被験者がもともと有していた疾患によってしばしばみられる症状であるような場合や併用薬剤の副作用として良く知られているような事象の場合には、被疑薬と有害事象の因果関係は確定的とは言えなくなる。こうした要因がある場合でも、個別の症例の基礎疾患の病状や併用薬剤の使用状況、有害事象の重症度や発現時期によっては被疑薬の因果関係を積極的に疑うことが合理的な場合もありうる。
また、有害事象の性質や、被疑薬(代謝物を含む)の類薬についての以下のような情報がもしあれば、これらも含め総合的に因果関係が評価される場合もある。
上述の情報を考慮しても、個別症例の有害事象が被疑薬と因果関係があるのか否かの判断が困難なケースが少なくない。
治験で報告される有害事象の取り扱い
臨床試験のうち、新医薬品等の承認を得るための臨床試験を治験と呼び、これはいわゆるGCP省令に基づいて行われ、治験届がなされている。治験では、個別症例の有害事象について、治験責任医師等がその因果関係判断を行う。治験責任医師等が当該有害事象と試験薬との因果関係を否定しないと、治験依頼者によって個別症例の副作用として報告等の必要な手続きが検討される。この場合試験が継続中であれば、規制当局および治験に参加している他施設への迅速報告が検討されるとともに、その重要度によっては試験の継続についても検討される。しかしながら、試験が終了し、集積検討をする段階では、個別症例についての因果関係評価に基づいてそのまま、「当該試験薬が当該副作用を引き起こす」というように医薬品が評価されるわけではない。むしろ逆である。Food and Drug Administration (FDA)では審査官(reviewer)向けのガイドブックが作成され公開されている。このFDAレビューワーガイダンスによると、有害事象の報告に責任のある治験責任医師(investigator)あるいは治験依頼者(新医薬品等の申請者としてapplicantの用語で登場する)が行った個別症例についての因果関係判断からは、あまり有用な情報が得られない、あるいは、無視するようにとも取れる記述がみられる。以下に因果関係評価についての記述をFDAのレビューワーガイダンスより引用する。(http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072974.pdf)
どうして個別症例の因果関係評価を無視するような記述になっているかと言うと、集積検討をする場合には個別症例の因果関係評価とは別の情報が加わるからである。どのような情報かについては、具体例を提示することで解説したい。以下に臨床試験に関する教科書であるStatistical Issues in Drug Developmentからの例を2つほど引用する(3)。元の教科書を参照していただければわかるが、これらの例は架空の例であり、過去に実施された治験についての記録ではない。しかし、本稿では過去に起きたような時制で文章を記述させていただくことをあらかじめお断りしておく。
これらの例で興味深いのは、個別症例の因果関係判断が、集積検討の結果覆る点である。集積検討で得られる情報で重要な情報は、比較対象であるプラセボ群との発現頻度についての情報である。お示ししたような例が論理的にはあり得るため、FDAでは医薬品の有害事象の因果関係判断を行うに当たって、治験責任医師や治験依頼者の行った個別症例の因果関係判断を必ずしもそのままでは受け入れず、7.1.1の死因の記述にある通り頻度を確認するようにしている。頻度が同程度であっても、実薬で早期に発現していないか、重症度が実薬で高くないか等が比較されることもある。
ここで、本稿の趣旨とははずれるが、もう一点強調しておきたい点がある。先の例では集積検討の結果、当初報告してきた治験担当医の因果関係評価は「誤り」となる。にもかかわらず、医薬品評価としては何の問題も生じない点である。開鍵後の集積検討の結果を知ることなしに、それまでの医学的な知識に基づいて、一定の合理的な判断をしている限り、結果として因果関係評価が誤りであったとしても、試験上は問題にはならないのである。筆者の知る限り、国内で試験に参加される先生の中には、こうした「結果として誤り」となることを恐れるあまりか、例1のような場合で、因果関係は絶対に否定しないお考えの先生方もおられる。そうした判断の考え方は例2のような場合には逆に結果として「誤り」となってしまう恐れがあることも考慮されておくべきであろう。治験責任医師らはご自身の知識と経験そして、目の前の患者さんの状況を鑑み、因果関係があるのかないのかを素直に医学的に判断して、それでも「結果として誤り」となることは避けられないものである。
また、臨床試験に参加される先生方のご心配は他にもあるようだ。「因果関係はないと思うが、治験責任医師らが因果関係を否定してしまうと、その事象が誰からも評価されることなく承認審査が行われるのではないか」と懸念され、因果関係を否定することに躊躇する先生方もおられる。これにも、誤解がある。個別症例の因果関係を「なし」とした場合、確かに試験継続中の他施設や規制当局への迅速な報告はなされないであろう。しかし、FDAレビューワーガイダンスに示した通り集積検討の段階では、因果関係が否定されている事象も含め、有害事象のリストに記載され、集計され、規制当局へ報告される。これは国内でも同様である。言い換えるならば個別症例の因果関係を否定したとしても、その有害事象の情報が闇に埋もれてしまう心配はない。
治験責任医師らは、自身の医学的知識と経験にもとづき、目の前の被験者の状況を鑑み、因果関係があるのかないのか、医学的に判断することに専念することが求められる役割であろう。
臨床試験の限界
残念ながら、前述の例のようにプラセボと比較して実薬で発現頻度の高いものを「実薬による」とするように明快な判断ができる有害事象は、条件が整った限られたケースのようである。プラセボの情報が不十分な場合では、治療対象疾患の経過中に起きることが知られている合併症としての事象の頻度や、一般人口中での発症頻度、類薬での発現頻度などを比較の対象として判断する場合もあるかもしれない。いずれにしても上市時に得られている医薬品の副作用情報は限られている。市販後に初めてわかるような安全性情報も少なからずある。開発段階での安全性情報が不十分となる要因を先ほどとは別の教科書から引用する(4)。
このリストの中の問題の多くは、治験では選択基準に合致する限られた被験者に限られた期間のみ投与され、一定の観察期間のみの情報が収集されることから、市販後に起きる状況が十分評価できないのである。この要因の中で異質な点が5番目の要因である。ここに記載されている社会的要求が安全性の情報とのトレードオフとするような考え方に抵抗感を示す先生方もおられるかもしれない。しかし、過度な社会的要求は治験を最低限の期間で、かつ最小の被験者数で進めるような圧力になりかねない因子の一つである。そうした圧力の有無にかかわらず、治験段階での安全性評価は限られた情報であるとの認識に基づいて、市販後に安全性を監視し続けることが臨床医には求められていると考えられる。
市販後の副作用報告の取り扱い
市販後の安全性情報にはSolicited とUnsolicitedな情報がある。 Solicitedとは試験や調査等登録患者を一定期間観察して、有害事象が発症しないかを観察する、つまり観察される集団があらかじめ定義されている安全性情報をいう。市販後の情報の報告数を見るとSolicitedな情報も一部にはあるものの、数として多いのは自発報告等のUnsolicitedな情報である。自発報告とは、副作用症例を経験された臨床現場の先生方が任意でご報告される、副作用報告のことである。自発報告は2つの大きな問題を抱えており信頼できる発現頻度を計算することができない。第一は薬物曝露状況つまり、どのくらいの被疑薬投与症例に被疑薬がどのくらいの期間投与されたのか、事象の発現頻度であるincidenceを計算する場合の分母にできる数字がない。分母にできる数字の代替として思いつくものに、出荷数量から推計できる使用患者数がある。これについては、ヨーロッパの規制当局であるEMEA のガイドライン案によると、「すでに市販されている医薬品で、自発報告された有害事象数あるいは有害反応数を分子に、販売数を分母にした報告率は、医薬品使用者での有害反応の発生率の推定値として提示すべきではない。」とあり、少なくともヨーロッパでは好ましくないことと考えられている。出荷数量は流通在庫や期限切れによる廃棄の問題や一人当たりの使用量の推計が、どの程度実臨床の状況を反映されているのかが不明であるといった問題がある。このため分母が不正確にならざるを得ない状況があり、発生率の推定値としては好ましくないとしているのであろう。
第二は、実際に起きている有害事象のうち一部しか報告されていない、つまりアンダーレポートの問題があり、incidenceを計算する場合の分子にできる数字は、実際に事象が起きている件数より小さいと推定できる。そのほかにもいくつかの重要な問題があり以下にリスト化する。
不確実な診断は副作用報告の深刻な問題の一つである。自発報告では通常ごく限られた情報が報告されてくることから、規制当局や製薬企業が診断を確認することは困難である。さらに、報告者が薬局薬剤師、患者やその家族等であった場合は、診断や診断根拠を報告者自身が十分把握していない場合もある。副作用を診断した医師が報告する場合であっても、報告医師の専門分野以外の副作用については診断が正確でない場合が考えられる。また、他の病院などからの転院患者を引き受けて、その後起きた副作用を報告するような場合、前医で治療されていた基礎疾患やその臨床経過についてのデータを十分引き継いでいない場合もある。さらに、自発報告では仮によく知られている絞絡因子が当該症例にあったとしても、報告されてこないかもしれない。
診断自体に困難な点がない場合でも報告事象名の選択が悩ましいケースがある。例えば、高齢者の多発性骨髄腫の患者に化学療法が行われ、その後発熱および下痢を発症した。経過中、腎不全となり、死亡した。このように一連の経過で複数の病態が観察された場合に、報告者が副作用として選択する事象名にはぶれが生じうる。より多くの種類の病態が起きるような場合はさらに事象名の選択は複雑になる。これとは別の問題として、新聞やテレビといったマスメディアでセンセーショナルに取り上げられた副作用については、過去の経験にさかのぼって報告するなどにより急に副作用報告件数が増えることが知られている。また、多くの患者に使用される医薬品は、薬物とは因果関係のない偶発症等の情報を含め、多くの有害事象が報告されてくることになる。
自発報告の集積検討の試み
臨床試験では開鍵後に集積検討をすることができるが、自発報告には分母にできる数字や比較対象にできるプラセボ群もない。かといって、集積検討が全くできない訳ではない。世の中で起きている医薬品の副作用について迅速にうかがい知るうえで、現状として最大の情報を蓄積しているのが市販後の自発報告を含む副作用データベースである。前項のような自発報告の欠点があることを把握したうえで、精度は落ちるものの大量のデータから科学的推論をすることは可能であるとされ、医薬品曝露情報(incidence の分母に相当する情報)に代わる何らかの指標を用いる手法がいくつか開発されている。原理のもとにある考え方は単純である。表に示した通り、安全性データベースの中で、興味の対象である医薬品Xについて、興味の対象である有害事象Aが何件報告されているかをn(X,A)と表現する(表 パネルA)。

医薬品XについてA以外のすべての有害事象(!Aとする。以下同じ)の件数n(X,!A)とのオッズ比であるn(X,A)/n(X,!A)を計算する。X以外のすべての医薬品についても同様のオッズ比n(!X,A)/n(!X,!A)を計算し、この2つのオッズ比を比較する、すなわち、n(!X,A)/n(!X,!A)に対するn(X,A)/n(X,!A)の比を見ることで、相対的に医薬品Xについて有害事象Aが多く報告されていないかを検出する。これらの数字の比をROR(reporting odds ratio)と呼ぶ。具体的な数字を用いて計算過程を例示するため、2004年の第一四半期の3か月間にFDAに報告された副作用が疑われた症例の報告件数を表のパネルBに示す。この例ではゲフィチニブを興味の対象である医薬品とし、また、興味の対象である副作用を間質性肺炎とした。この3か月間にゲフィチニブが被疑薬として報告された間質性肺炎は22件あり、それ以外の報告事象は159件であった。ゲフィチニブについて、全有害事象報告件数に対する間質性肺炎の報告件数の比は、約0.138であった(パネルC-(1))。これに対して、同じ期間にゲフィチニブ以外の医薬品が被疑薬として報告された間質性肺炎は180件、ゲフィチニブ以外の医薬品が被疑薬として報告された間質性肺炎以外の事象は49983件であった。ゲフィチニブ以外の医薬品について、全有害事象報告件数に対する間質性肺炎の報告件数の比は、約0.00360であった(パネルC-(2))。ゲフィチニブのROR値は0.138と0.00360の比である、約38.3となる。ゲフィチニブについて、それ以外の医薬品と同程度の間質性肺炎が報告されてくると仮定すると期待されるROR値(帰無仮説H0 に基づくROR値)は1.00であるのに対し、計算結果は約38.3であった。閾値をどうするかという議論はあるが、この値は一般的にはシグナルと判断できる程度に大きい値である。ここで、原理を考えていただくためにお示ししたRORは、データベースに入力されている件数が少ない事象や被疑薬としての報告件数が少ない医薬品についての精度が不十分であるとの指摘もある。その精度を上げるべく開発されているものがいくつかある。「はずれ値」を検出するという目的から、neural networkやsupport vector machineといったアルゴリズムを応用することも検討されているが、本稿ではそうしたデータマイニング手法の一つを次の例としてお示しする。それは世界保健機構(World Health Organization, WHO)が開発したBayesian Confidence Propagating Neural Network (BCPNN) という手法であり、BCPNNを用いてFDAが公開しているAdverse Event Reporting System (AERS)データベースのデータを集計し、一般によく知られているいくつかの副作用等を継時的に表現し簡単な解説を加える。
本稿ではBCPNNの計算結果出力されるInformation Components (IC)値およびstandard deviation (SD) 値を示すが、BCPNNの計算方法や解釈についての詳細は他論文を参照していただきたい(5)。FDA AERSのデータを2004年第1四半期から2010年第2四半期まで過去に報告した方法に従い入手した(1)。四半期ごとにそれまでの集積状況をもとに計算した結果を表示したのが図1である。スタチンとして集計した医薬品には、simvastatin, atorvastatin, rosuvastatin, pravastatin, fluvastatin, losuvastatin, cerivastatin and pitavastatinが含まれる。また、アンジオテンシン変換酵素(ACE)阻害薬には、captopril, enalapril, alacepril, delapril, cilazapril, lisinopril, benazepril, imidapril, temocapril, trandolapril, perindoprilが含まれる。そしてARBには、olmesartan, telmisartan, valsartan, candesartan, irbesartan, losartanが含まれる。プロットされている点は2004年第1 四半期から各報告期間までに集積された累積のIC値である。エラーバーはSD の2倍である2SD が表示されている。BCPNNの開発時に過去の事例でシグナルの検出を試した結果、IC-2SD > 0 すなわち、図で言えばエラーバーの下限が0のラインより上に来たらシグナルとみなすこととされている。本稿の例ではゲフィチニブの間質性肺炎、スタチンの横紋筋融解症、ACE阻害薬の咳についてのIC値は、いずれの時点で見てもそれらのエラーバーの下限が0のラインを上回っている。言い換えるならば、BCPNN法を用いてFDAのデータを解析すると、医薬品リスクのシグナルが検出されたことになる。これに対し、ARBの横紋筋融解症についてのIC値は解析した期間中にエラーバーの下限が0のラインを上回っているポイントはなく、BCPNN法ではシグナルは検出されない。つまり、BCPNNのように相対的な報告頻度という観点からの集計では、ARBによって横紋筋融解症が起こりかねないと懸念する根拠を見出すことはできなかった。
なお、FDAのデータは日本からの報告も含まれるが、間質性肺炎等日本からの報告が多い一部の例を除くと、日本よりは米国での発現状況を色濃く反映していると考えられる。つまり、日本国内での副作用発現状況については、FDA AERSとは異なる可能性は否定できない。国内の副作用状況はPMDAが医薬品ごとの副作用情報を公開している。しかし全医薬品についての報告数を集計する必要のあるBCPNN等のデータマイニング手法は、PMDAの外部の研究者にとって現実的には難しい。本稿では紹介しなかったが、イギリスの規制当局で採用されているProportional Reporting Ratio (PRR)と言われる手法や、FDAで採用されているGamma Poisson Shrinker program (GPS)の手法も基本的にはBCPNNと同様のデータを用いて計算される。いずれも、大量のデータに埋もれている問題を拾い出す、新たな問題の提起に有用と考えられているが、検証的な評価には必ずしも向いていないとされる。
さいごに
ある患者に治療を行い、その後治ったとする。その患者さんにとっては良いことかもしれないが、個別の症例を見ている限り、その治療が本当に効いたのかどうかについて判断することは困難である。同様に個別症例に治療を行い、その後有害事象が起きた。臨床経過を見るとその個別症例にとって医薬品による副作用と考えられる場合でも、その医薬品と有害事象の因果関係を判断することは困難である。臨床試験で検出できる医薬品のリスクも限界がある。本稿では個別症例および集積検討を行う場合の有害事象の医薬品との因果関係評価の考え方や、市販後副作用データベースを利用し、限られた現在の状況で医薬品の副作用リスクのシグナルを検出する方法等について紹介した。本稿で紹介したBCPNN等ではシグナルが検出されない医薬品リスクもあるだろう。冒頭のARBの例では、副作用として報告した臨床家の先生方、専門協議にご参加の先生方それぞれが、当該症例の報告について詳細な情報を検討され、当該医薬品について真摯にリスクとベネフィットをお考えの上で行動された結果が使用上の注意の改訂指示につながっていると理解している。使用上の注意の改訂等のリスクコミュニケーションは一般に、検出されたシグナルがはっきりリスクであったと確認される頃に発出されても被害が甚大となり、手遅れと言われかねないことを考えると、コンサーバティブな方向に偏るのもやむを得ないことと思われる。リスクコミュニケーションの結果、社会全体で救われる患者さんがわずかでもおられれば良いのかもしれない。一方でリスクコミュニケーションが過剰であったならば、必要な治療を受ける機会が狭められる患者が出る懸念や、リスクコミュニケーション自体の信頼性が低下しかねない懸念についても思いをはせる必要がある。情報の受け手である医師は提供された情報が、どの程度のリスクなのか、どのような情報に基づいて判断されたのかについて、その根拠を吟味し、科学的な視座より把握した上で日常の診療へ結び付ける努力が求められる。
著者のCOI開示:
報酬(サノフィ・アベンティス株式会社)
Figure Legend
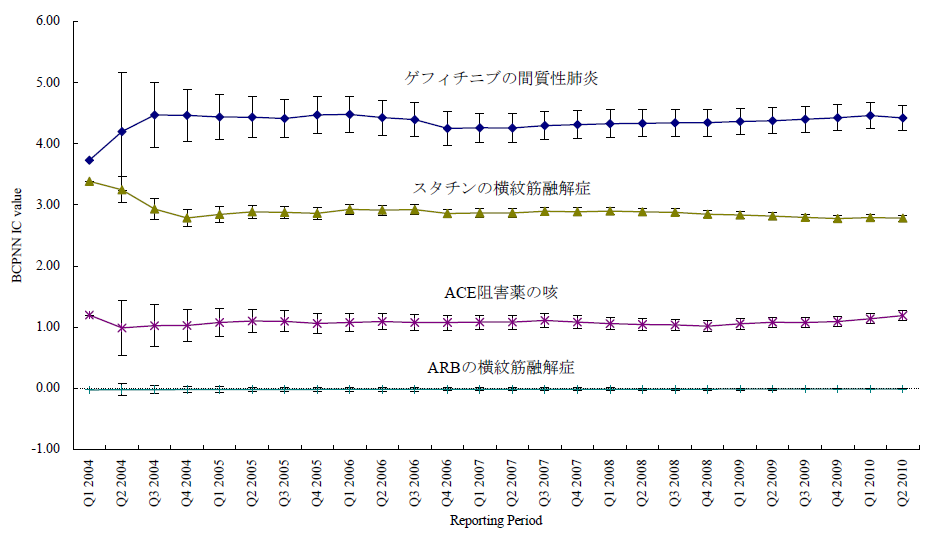
図1 報告期間とBCPNN IC値
ARBの横紋筋融解症および、副作用としてよく知られているゲフィチニブの間質性肺炎、スタチンの横紋筋融解症、ACE阻害薬の咳について四半期ごとに区切った報告期間までの累積BCPNN IC をプロットした。エラーバーは2SDを示す。よく知られた3つの副作用の例ではエラーバーの下限が0を超えておりBCPNN法でシグナルが検出された。一方、ARBの横紋筋融解症についてはエラーバーの下限が0を超える期間はなく、シグナルはBCPNN法で検出されなかった。
文献
- Oshima Y: Characteristics of drug-associated rhabdomyolysis: analysis of 8,610 cases reported to the u.s. Food and drug administration. Intern Med 50: 845-53, 2011.
- USE ICOHOTRFROPFH: http://www.ich.org/. In: ICH HARMONISED TRIPARTITE GUIDELINECLINICAL SAFETY DATA MANAGEMENT. 1994.
- Senn S: Statistical Issues in Drug Development. In: John Wiley & Sons, Ltd, 2007.
- Spilker B: Guide to Clinical Trials. In: Lippincott Williams & Wilkins, 1991.
- Bate A, Lindquist M, Edwards IR, Olsson S, Orre R, Lansner A and De Freitas RM: A Bayesian neural network method for adverse drug reaction signal generation. Eur J Clin Pharmacol 54: 315-21, 1998.
被偽薬ではなく、被疑薬です。
ご参考ください。