




遺伝性白質疾患ガイドライン
アレキサンダー病
Alexander disease(ALXDRD,OMIM#203450)
疾患説明; アストロサイト細胞質内のローゼンタル線維を病理学的特徴とする中枢神経系の白質変性疾患.98%の症例でglial fibrillary acidic protein (GFAP)をコードするGFAP遺伝子に変異を認める.発症年齢は乳児期から高齢者まで幅広い.臨床病型は主に乳児期発症で大脳白質病変を主体とする型(大脳優位型:1型), 主に成人期発症で延髄・脊髄病変を主体とする型(延髄・脊髄優位型:2型)および両者の特徴をもつ型(中間型:3型)に分類される.
治療; 根治治療法はなく,抗けいれん薬などによる対症療法が行われる.
I. 概要
1. 定義
1949年にAlexander WSが記載した難治性けいれん,水頭症,精神遅滞を呈した15か月の乳児剖検例が最初の報告である1).病理学的にアストロサイトの細胞質内にグリア線維性酸性蛋白(glial fibrillary acidic protein: GFAP),αB-クリスタリン,熱ショック蛋白を主な構成成分とするローゼンタル線維を認めることが特徴であり,98%の症例においてGFAP遺伝子変異を認める2).
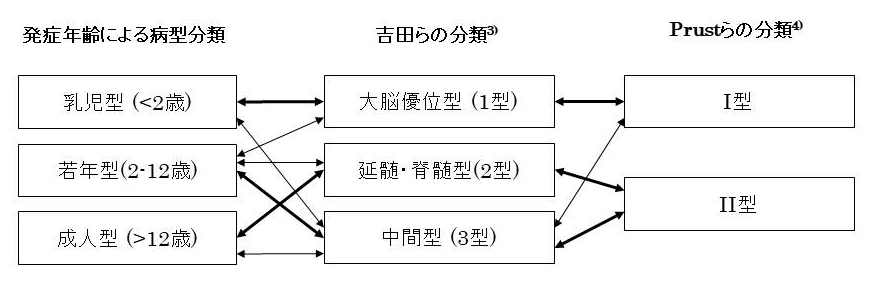
臨床病型は乳児型(2歳未満の発症),若年型(2歳から12歳未満の発症)および成人型(12歳以上の発症)の発症年齢による分類が用いられてきたが,2011年にPrustら3)と吉田ら4)は独立して新しい病型分類を提唱した.
① Prustらの病型分類3)
既報告185症例と新規の30症例に対して統計学的手法を用いて以下の2型に分類した.
I型:早期発症でけいれん,大頭症,運動発達遅延,脳症,栄養不良,発作性の症状悪化を認め,典型的な頭部MRI所見5)を呈する.
II型:晩期発症で自律神経障害,眼球運動障害,球症状を認め,非典型的な頭部MRI所見6)を呈する.
② 吉田らの病型分類4)
日本の全国調査にて得られた35症例の臨床症状およびMRI画像所見に基づいて以下の3型に分類した.
大脳優位型(1型):けいれん,大頭症,精神運動発達遅滞を認め,頭部MRIにて前頭部優位の大脳白質病変を認めることが特徴である.主に乳幼児期発症で,機能予後不良の重症例が多い.新生児期発症例では水頭症や頭蓋内圧亢進症状をきたし,生命予後不良である.
延髄・脊髄優位型(2型):筋力低下,痙性麻痺,球麻痺/仮性球麻痺,運動失調,自律神経障害などを種々の組み合わせで認め,MRIにて延髄・上位頚髄の信号異常あるいは萎縮を認めることが特徴である.学童期から成人期以降の発症で,他の病型と比較して緩徐な経過をとることが多い.
中間型(3型):1型および2型の両者の特徴を有する.発症時期は幼児期から成人期まで幅広い.
乳児型の発症年齢および臨床所見はPrustらのI型および吉田らの1型にほぼ対応し,成人型の臨床所見はPrustらのII型および吉田らの2型あるいは3型にほぼ対応する.大脳症状と延髄・脊髄症状の両者を示す症例は吉田らの3型に分類できるが,他の分類ではそれぞれのすべての病型に該当しうる(図1).なお,本ガイドラインの各項目は各参考文献で使用された分類に従って記載した.
図1 アレキサンダー病の病型分類ならびに各分類の関連
2) 疫学
頻度:日本における全国調査から有病者数は約50名,有病率は270万人に1人と推定される4).
性差:乳児型では男児優位の傾向があるが,若年型・成人型では男女差を認めない4).
病型別頻度:GeneRevies®による既報告をもとに算出した病型別頻度は乳児型が約42%,若年型が約22%,成人型が約33%である7).日本の全国調査から算出した病型別頻度は乳児型が27.3%,若年型が24.2%,成人型が48.5%であった4).
遺伝:乳児型および若年型はほぼすべてがde novo変異である4).成人型の約65%で常染色体優性遺伝形式を示唆する家族内発症が認められた4).しかし,家系解析の報告は少なく,浸透率についても不明である.
3) 病因・病態
ヒト野生型GFAPを過剰発現させたトランスジェニックマウスにおいてGFAP発現量に比例した寿命の短縮とローゼンタル線維の出現を認めた8)ことから機能獲得性機序が推定されている.一方,アレキサンダー病患者ではGFAP遺伝子変異(大部分がミスセンス変異)を認めるが2),変異GFAP遺伝子導入トランスジェニックおよびノックインモデルマウスは臨床表現型を十分に示さない9),10).このことから,GFAP遺伝子変異はアレキサンダー病発症の必要条件であるが,GFAP発現量の増加を十分条件とすることが示唆される.
現段階で提唱されている病態仮説は以下の通りである11)-13).GFAP遺伝子変異に伴い形成される”変異GFAPオリゴマー(4-6分子のcoiled-coil重合体)”によりプロテアソーム系の機能が低下する.これにより毒性閾値を超える変異GFAPの蓄積とアストロサイトの活性化が生じ,変異GFAPの合成がさらに促進されるポジティブフィードバックが形成される.変異GFAPの蓄積が促進されるにつれて,分子シャペロンであるαB クリスタリンの枯渇やグルタミン酸トランスポーターの異常などのアストロサイト機能障害,ローゼンタル線維の形成がもたらされる.
しかし,GFAP合成促進に関わるポジティブフィードバックやローゼンタル線維の毒性効果およびアストロサイト機能障害の詳細については十分な証明がされていない.
なお,ヒトのアレキサンダー病においてGFAP遺伝子のmultiplicationの報告はなく,GFAPの量的変化に影響を与える修飾因子の存在が示唆される.遺伝的修飾因子の候補の1つとしてGFAPプロモーター遺伝子多型が報告されている14).
アレキサンダー病のもう一つの病理学的特徴である脱髄については,モデルマウスの研究からK緩衝系の異常によるミエリン形成や維持の障害が推測されている15)が詳細な機序は不明である.
4) 臨床症状
新生児から70歳代まで幅広い年齢層で発症が認められる.
1型:けいれん,大頭症,精神運動発達遅滞が3大症状である4).けいれんは難治性のことが多いが,学童期以降に軽減する症例もある.大頭症は乳児期から学童期に目立つが,成人になるにつれて目立たなくなる.経過とともに痙性麻痺,構音障害,発声障害,嚥下障害などの延髄・脊髄症状が進行する.新生児期発症例は水頭症,頭蓋内圧亢進,難治性けいれんおよび発育不良をきたし,重症である.
2型:四肢筋力低下,痙性麻痺,筋強剛, 四肢・体幹失調,構音障害,発声障害,嚥下障害,自律神経障害(起立性低血圧,膀胱直腸障害,睡眠時無呼吸)などの延髄・脊髄症状および小脳症状を種々の程度で認める4).運動症状はしばしば左右差を認める.口蓋振戦は高頻度ではないが家族性の場合はアレキサンダー病が強く疑われる.1型の3大症状であるけいれん,大頭症,精神運動発達遅滞は通常認めない.認知症の報告は少数であるが,前頭側頭型認知症に類似した認知機能低下を示した症例が存在する.一過性の反復性嘔吐が唯一の症状で,MRIにて両側延髄背側に結節状病変を示す小児の症例報告がある16),17).
3型:1型と2型の両者の特徴を有する4).1型の長期生存例,精神運動発達遅滞+2型が含まれる.後者の症例は学童期以降に2型の症状が顕在化して初めて医療機関を受診することもあり,その際に精神運動発達遅滞が判明することがある.また,熱性けいれんや原因不明の脳症の既往をもつ症例もあり,乳幼児期にけいれんを繰り返していたが,学童期までに軽減あるいは消失したという病歴が聴取できることがある.側彎などの脊柱異常や複視を伴うことがある.
5) 検査
5)-1 画像検査
1型:van der Knaapらは白質脳症患者217名に対する頭部MRIの多施設後ろ向き研究にて,①前頭部優位の広範な大脳白質異常,②T2強調画像で低信号,T1強調画像で高信号を示す脳室周囲の縁取り,③基底核と視床の異常,④脳幹の異常(特に中脳と延髄),⑤造影効果 (脳室周囲,前頭葉白質,視交叉,脳弓,基底核,視床,小脳歯状核,脳幹の1か所以上)の5項目の基準のうち4項目を満たした場合はアレキサンダー病の病理学的所見と一致率が高いことを報告した5).
2型:頭部MRIにて延髄・頚髄の萎縮・異常信号をほぼ全例で認める4),/.典型的には橋が保たれた延髄・上位頚髄の著明な萎縮を呈するオタマジャクシ様(tadpole appearance)の脳幹形態を示す18).高齢者や軽症例で延髄・頚髄の萎縮が軽度の場合は頭部MRI水平断にてメダマチョウの眼状紋(eye spot)様の延髄錐体の異常信号を認めることがある19).大脳,中脳,橋の錘体路には通常異常信号を認めない.10歳代前半から20歳代の若年例では延髄の結節状・腫瘤状異常信号を認めることがある.小脳歯状核門の信号異常20)やFLAIR像にて中脳の縁取り(midbrain periventricular rim) 21)も高率にみられる所見である.
テント上はT2強調画像にてperiventricular garlandと表現される側脳室壁に沿った花弁状の高信号を認める6).この病変は造影効果を示すことがあり,ローゼンタル線維が多数出現する部分とされる.前頭葉優位の大脳白質病変は認めないが,側脳室前角周囲にcap状病変を示すことがある.
3型:大脳白質病変と延髄・頚髄の萎縮・異常信号といった1型と2型の両者の特徴を呈する4).大脳白質病変の程度には症例差があり,嚢胞化を伴う症例が多い.Megalencephalic leukoencephalopathy with subcortical cystsやvanishing white matter diseaseと鑑別を要するが,3型アレキサンダー病では延髄・頚髄萎縮が必発であることが鑑別点である.
5)-2 遺伝子検査
98%の症例においてGFAP遺伝子変異を認める2),7).これまで100種類以上のGFAP遺伝子変異が報告されている.大多数がミスセンス変異であるが,インフレーム挿入/欠失変異,終止コドン近傍のフレームシフト変異およびスプライス変異の報告もある22).ミスセンス変異はGFAPドメインの全域に分布するが,rod domainの1A, 2A, 2B領域とtail domainに多く,特にCpGが関与するR79,R88,R239,R416が置換される変異は人種を越えて認められ,アレキサンダー病のhot spotである2).R79,R88,R239が置換される変異は1型および3型で認められ,R416が置換される変異(R416W)は1型,2型,3型すべての型で報告されている4).一方,2型に頻度の高い変異は特に存在しない4).
6) 治療およびケア
治療は対症療法にとどまる.けいれんに対する抗てんかん薬の投与,栄養管理,併発する感染症に対する抗生物質の投与,痙性麻痺に対する抗痙縮薬,学習障害や認知機能障害に対する療育・ケアが行われる7).
7) 食事・栄養
栄養管理,併発する感染症に対する抗生物質の投与,学習障害や認知機能障害に対する療育を行う7).運動機能障害として筋力低下,痙性麻痺,四肢・体幹失調を認めうるが,症状の程度や組み合わせには個人差があり,適切な評価のうえでのリハビリテーションが望まれる.嚥下障害がみられる場合は,とろみ食にするなど食事形態の工夫が必要である7).
8) 予後
I型の生命予後は約14年である3).新生児期発症例は水頭症や頭蓋内圧亢進症状のために生後数週から数か月で死亡することが多い.乳児期発症例は難治性けいれんや栄養障害,感染症などのため学童期までに死亡する症例が多い.一方で学童期までにけいれんが軽減・消失する症例も存在し,このような症例は精神遅滞と進行性の歩行障害や嚥下障害など延髄・脊髄症状を認めるものの生命予後は新生児期発症例や難治性けいれんを伴う乳児期発症例と比較すると良好である.II型の生命予後は約25年3)でありI型と比較して良好であるが,運動症状・球症状・呼吸症状などが急激に増悪する症例23)から軽微な症状24)にとどまる症例まで機能予後としては症例間の差が大きいことが推測される.
II. 診断基準
A. 神経症状
- けいれん
- 大頭症
- 精神運動発達遅滞
- 四肢運動障害:筋力低下,痙性麻痺,小脳性運動失調,筋強剛
- 球麻痺あるいは仮性球麻痺:嚥下障害,構音障害,発声障害
- 自律神経障害:起立性低血圧,膀胱直腸障害,睡眠時無呼吸
- 口蓋振戦
- 反復性嘔吐
B. MRI所見
- 前頭部優位の大脳白質信号異常
- 脳室周囲の縁取り;T2強調画像で低信号,T1強調画像で高信号を示す
- 基底核と視床の異常;T2強調画像で高信号を伴う腫脹または高・低信号を伴う萎縮
- 造影効果;脳室周囲,前頭葉白質,視交叉,脳弓,基底核,視床,小脳歯状核,脳幹など
- 脳幹の異常・萎縮
1) 中脳の信号異常
2) 延髄・上位頚髄の異常.
a) 橋底部が保たれ,延髄および上位頚髄が萎縮する像
b) T2強調画像における延髄錐体や頚髄の信号異常
c) 萎縮を伴わない結節性腫瘤像 - 小脳歯状核門の信号異常あるいは萎縮
C. 遺伝子検査および病理学的検査
- 遺伝子検査:GFAP遺伝子変異を同定
- 病理学的検査:大脳白質,上衣下および軟膜下のアストロサイト細胞質内に特徴的なローゼンタル線維を認める
D. 鑑別診断
Pelizaeus-Merzbacher病をはじめとする先天性大脳白質形成不全症,megalencephalic leukoencephalopathy with subcortical cysts, 副腎白質ジストロフィー, 異染性白質ジストロフィー, メロシン欠損型先天性筋ジストロフィー, Krabbe病, vanishing white matter disease, Canavan病, 脳腱黄色腫, 多発性硬化症, neuromyelitis optica, 急性散在性脳脊髄炎, 進行性多巣性白質脳症, 脳腫瘍, 脳血管障害, CADASIL, CARASIL, ミトコンドリア脳筋症, 遺伝性痙性対麻痺, HTLV-I関連脊髄症, ALSなど大脳白質や延髄・脊髄に病変の主座を認める疾患
確定診断:
Definite:
①A.の1.~3.の1項目以上,およびB.の1.の所見を認める
②A.の4.~8.の1項目以上,およびB.の5.の2).に挙げる項目の1つ以上の所見を認める
上記の①あるいは②を満たし,C.の1.あるいは2.を認めた場合
Probable:
①A.の1.~3.の1項目以上,およびB.の1.~5.のうち4つ以上の所見を認める
②A.の4.~8.の1項目以上,およびB.の5.の2).に挙げる項目の1つ以上および6.の所見を認める
上記の①あるいは②を満たし,D.の鑑別診断を除外できた場合
DefiniteおよびProbableの①は吉田らの1型,②は2型,①および②の両者を満たすものは3型に相当する.
III. 最近のトピック
病態仮説に基づき,GFAPの発現抑制やαBクリスタリンの発現増加をもたらす薬剤が治療候補薬剤としていくつか報告されている.細胞実験レベルでは第3世代セフェム系の抗生剤であるceftriaxone25)やポリフェノール類の1つであるcurcumin26)の報告がある.モデルマウスにおいては抗精神病薬のリチウムの有効性が報告されている27).ただし,治療有効域が狭く毒性も強いことが問題点とされる27).別の治療戦略として,グリア由来のNOが非細胞自律性神経変性に関与することを示した研究28)やムスカリン性アセチルコリン受容体アンタゴニストによる酸化ストレス抑制に注目した研究がある29).さらにアレキサンダー病患者の皮膚あるいは血液から誘導したiPS細胞の作製が報告され30),今後の病態および治療研究の重要なツールとなることが期待される.
引用文献 (末尾に記載のないものは全てエビデンスレベルVI)
- Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 1949: 72: 373-381.エビデンスレベル V
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 2001: 27: 117-120. エビデンスレベル IVb
- Prust M, Wang J, Morizono H, Messing A, Brenner M, Gordon E, Hartka T, Sokohl A, Schiffmann R, Gordish-Dressman H, Albin R, Amartino H, Brockman K, Dinopoulos A, Dotti MT, Fain D, Fernandez R, Ferreira J, Fleming J, Gill D, Griebel M, Heilstedt H, Kaplan P, Lewis D, Nakagawa M, Pedersen R, Reddy A, Sawaishi Y, Schneider M, Sherr E, Takiyama Y, Wakabayashi K, Gorospe JR, Vanderver A. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 2011: 77: 1287-1294. エビデンスレベル IVb
- Yoshida T, Sasaki M, Yoshida M, Namekawa M, Okamoto Y, Tsujino S, Sasayama H, Mizuta I, Nakagawa M. Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J Neurol 2011; 258: 1998-2008. エビデンスレベル IVb
- van der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S, Begger JC, van Coster R, Barth PG, Thomas NH, Valk J, Powers JM. Alexander disease: diagnosis with MR imaging. Am J Neuroradiol 2001: 22: 541-552 エビデンスレベル IVb
- van der Knaap MS, Ramesh V, Schiffmann R, Blaser S, Kyllerman M, Gholkar A, Ellison DW, van der Voorn JP, van Dooren SJM, Jakobs C, Barkhof F, Salomons GS. Alexander disease. Ventricular garlands and abnormalities of the medulla and spinal cord. Neurology 2006: 66: 494-498 エビデンスレベル V
- Srivastava S,Naidu S. Alexander disease. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. 2002 Nov 15 [updated 2015 Jan 8].
- Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol 1998: 152: 391-398.
- Hagemann TL, Connor JX, Messing A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and white matter stress response. J Neurosci 2006: 26: 11162-11173.
- Tanaka FK, Takebayashi H, Yamazaki Y, Ono K, Naruse M, Iwasato T, Itohara S, Kato H, Ikenaka K. Murine model of Alexander disease: analysis of GFAP aggregate formation and its pathological significance. GLIA 2007: 55: 617-631.
- Tang G, Xu Z, Goldman JE. Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem 2006: 281: 38634-38643.
- Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE. Oligomers of mutant glial fibrillary acidic protein (GFAP) inhibit the proteasome system in Alexander disease astrocytes, and the small heat shock protein αB-crystallin reverses the inhibition. J Biol Chem 2010: 285: 10527-10537.
- Hagemann TL, Boelens WC, Wawrousek EF, Messing A. Suppression of GFAP toxicity by B-crystallin in mouse models of Alexander disease. Hum Mol Genet 2009: 18: 1190-1199.
- Yoshida T, Mizuta I, Saito K, Ohara R, Kurisaki H, Ohnari K, Riku Y, Hayashi Y, Suzuki H, Shii H, Fujiwara Y, Yonezu T, Nagaishi A, Nakagawa M. Effects of a polymorphism in the GFAP promoter on the age of onset and ambulatory disability in late-onset Alexander disease. J Hum Genet 2013: 58: 635-638. エビデンスレベル V
- Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE. Alexander disease. J Neurosci. 2012: 32: 5017-5023.
- Niinikoski H, Haataja L, Brander A, Valanne L, Blaser S. Alexander disease as a cause of nocturnal vomiting in a 7-year-old girl. Pediatr Radiol 2009: 39: 872-875. エビデンスレベル V
- Namekawa M, Takiyama Y, Honda J, Sakoe K, Naoi T, Shimazaki H, Yamagata T, Momoi MY, Nakano I. A novel adult case of juvenile-onset Alexander disease: complete remission of neurological symptoms for over 12 years, despite insidiously progressive cervicomedullary atrophy. Neurol Sci 2012: 33: 1389-1392. エビデンスレベル V
- Namekawa M, Takiyama Y, Honda J, Shimazaki H, Sakoe K, Nakano I. Adult-onset Alexander disease with typical “tadpole” brainstem atrophy and unusual bilateral basal ganglia involvement: a case report and review of the literature. BMC Neurol 2010: 10:21. エビデンスレベル V
- Yoshida T, Mizuta I, Saito K, Kimura Y, Park K, Ito Y, Haji S, Nakagawa M, Mizuno T. Characteristic abnormal signals in medulla oblongata-‘eye spot’ sign: four cases of elderly-onset Alexander disease. Neurology clinical practice 2015: 5: 259-262. エビデンスレベル V
- Graff-Radford J, Schwartz K, Gavrilova RH, Lachance DH, Kumar N. Neuroimaging and clinical features in type II (late-onset) Alexander disease. Neurology 2014: 82: 49-56. エビデンスレベル V
- Farina L, Pareyson D, Minati L, Ceccherini I, Chiapparini L, Romano S, Gambaro P, Fancellu R, Savoiardo M. Can MR imaging diagnose adult-onset Alexander disease? Am J Neuroradiology 2008: 29: 1190-1196. エビデンスレベル V
- Flint D, Li R, Webster LS, Naidu S, Kolodny E, Percy A, van der Knaap M, Powers JM, Mantovani JF, Ekstein J, Goldman JE, Messing A, Brenner M. Splice site, frameshift, and chimeric GFAP mutations in Alexander disease. Hum Mutat 2012: 33: 1141-1148. エビデンスレベル V
- Ayaki T, Shinohara M, Tatsumi S, Namekawa M, Yamamoto T. A case of sporadic adult Alexander disease presenting with acute onset, remission and relapse. J Neurol Neurosurg Psychiatry 2010: 81: 1292-1293. エビデンスレベル V
- Sugiyama A, Sawai S, Ito S, Mukai H, Beppu M, Yoshida T, Kuwabara S. Incidental diagnosis of an asymptomatic adult-onset Alexander disease by brain magnetic resonance imaging for preoperative evaluation. J Neurol Sci 2015: 354: 131-132. エビデンスレベル V
- Bachetti T, Zanni ED, Balbi P, Bocca P, Prigione I, Deiana GA, Rezzani A, Ceccherini I, Sechi GP. In vitro treatments with ceftriaxone promote elimination of mutant glial fibrillary acidic protein and transcription down-regulation. Exp Cell Res 2010: 316: 2152-2165.
- Bachetti T, Di Zanni E, Balbi P, Ravazzolo R, Sechi GP, Ceccherini I. Beneficial effects of curcumin on GFAP filament organization and down-regulation of GFAP expression in an in vitro model of Alexander disease. Exp Cell Res 2012: 318: 1844-1854.
- LaPash Daniels CM, Paffenroth E, Austin EV, Glebov K, Lewis D, Walter J, Messing A. Lithium decreases glial fibrillary acidic protein in a mouse model of Alexander disease. PLoS One 2015: 10: e0138132.
- Wang L, Hagemann TL, Kalwa H, Michel T, Messing A, Feany MB. Nitric oxide mediates glial-induced neurodegeneration in Alexander disease. Nat Commun 2015: 6:8966.
- Wang L, Hagemann TL, Messing A, Feany MB. An in vivo pharmacological screen identifies cholinergic signalling as a therapeutic target in glial-based nervous system disease. J Neurosci 2016: 36: 1445-1455.
- Kondo T, Funayama M, Miyake M, Tsukita K, Era T, Osaka H, Ayaki T, Takahashi R, Inoue H. Modeling Alexander disease with patient iPSCs reveals cellular and molecular pathology of astrocytes. Acta Neuropathol Comm 2016: 4:69.
文献検索
1. 概要
1)定義
- PubMed(検索2016年12月27日)
“Alexander Disease”[MeSH Major Topic] 195件 - 医中誌(検索2016年12月27日)
((Alexander病/TH or アレキサンダー病/AL)) and (PT=会議録除く) 88件
2)疫学
- PubMed(検索2016年12月27日)
“Alexander Disease/epidemiology”[Mesh] 4件 - 医中誌(検索2016年12月27日)
((Alexander病/TH or アレキサンダー病/AL)) and((有病率/TH or 有病/AL))and (PT=会議録除く) 1件
3)病因・病態
- PubMed(検索2016年12月27日)
“Alexander Disease/etiology”[Majr] 106件 - 医中誌(検索2016年12月27日)
((病態生理/TH or 病態/AL))or病因/AL)and ((Alexander病/TH or アレキサンダー病/AL)) and (PT=会議録除く) 21件 - ハンドリサーチ
4)臨床症状
- PubMed(検索2016年12月27日)
“Alexander Disease”[Majr] AND “humans”[MeSH Terms] 184件 - 医中誌(検索2016年12月27日)
((Alexander病/TH or アレキサンダー病/AL)) and((徴候と症状/TH or 症状/AL)) and (PT=会議録除く) 17件
5)検査
5)-1 画像
5)-2 遺伝子検査
6) 治療・ケア
7)食事・栄養
- PubMed(検索2016年12月27日)
“Alexander Disease”[MeSH Major Topic] 195件 - 医中誌(検索2016年12月27日)
((Alexander病/TH or アレキサンダー病/AL)) and (PT=会議録除く) 88件
8)予後
- PubMed(検索2016年12月27日)
((“prognosis”) OR “mortality”) AND “Alexander Disease”[MeSH Major Topic] 5件 - 医中誌(検索2016年12月27日)
((Alexander病/TH or アレキサンダー病/AL)) and((予後/TH or 予後/AL))and (PT=会議録除く) 2件
3. 最近のトピック
- PubMed(検索2016年12月27日)
“Alexander Disease”[Majr] AND (“2014/01/01”[PDAT] : “2016/12/31”[PDAT]) 28件 - 医中誌(検索2016年12月27日)
((Alexander病/TH or アレキサンダー病/AL)) and (DT=2011:2016 PT=会議録除く) 24件









