








Guidelines on Hereditary Leukodystrophies
Alexander disease (ALXDRD; OMIM #203450)
Disease description: Alexander disease (ALXDRD) is a degenerative disease of the white matter in the central nervous system that is pathologically characterized by Rosenthal fibers within the astrocyte cytoplasm. In 98% of the affected patients, a mutation is present in the gene encoding glial fibrillary acidic protein (GFAP). ALXDRD occurs in a wide range of age groups, from infancy to old age, and is classified into three clinical types: type 1 (cerebral), which occurs in infancy and is characterized by predominantly cerebral white-matter lesions; type 2 (bulbospinal), which mainly develops in adulthood and is characterized by lesions predominantly in the medulla oblongata and spinal cord; and type 3 (intermediate), which combines characteristics of both.
Treatment: There is no cure for ALXDRD; rather, symptomatic therapy with anti-epileptics and other drugs is provided.
I. Overview
1. Definition
Alexander disease (ALXDRD) disease was first described in 1949 by Alexander, who reported the autopsy findings of a 15-month-old infant with intractable seizures, hydrocephalus, and intellectual disability.1 Pathological tests reveals characteristic Rosenthal fibers consisting mainly of glial fibrillary acidic protein (GFAP), ab-crystallin, and heat shock protein in the cytoplasm of astrocytes; 98% of affected patients carry a mutation in the GFAP gene.2)
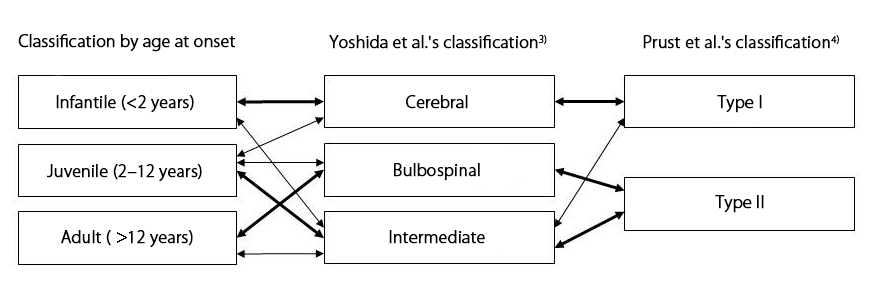
Although conventionally it has been classified clinically as infantile (onset before age 2 years), juvenile (onset between age 2–11 years), and adult (onset at age 12 years or older), in 2011 Prust et al.3) and Yoshida et al.4) independently proposed new pathological classifications.
- Prust et al.'s pathological classification3)
Prust et al. proposed a two-type classification based on a statistical study of 185 previously reported cases and 30 new cases.
Type I: Early-onset with seizures, macrocephaly, delayed motor development, encephalopathy, malnutrition, and episodes of symptom aggravation, with typical signs on cranial magnetic resonance imaging (MRI).5)
Type II: Late-onset with dysautonomia, oculomotor disturbance, and bulbar symptoms, with atypical signs on cranial MRI.6) - Yoshida et al.'s pathological classification4)
Yoshida et al. proposed a three-type classification based on data on the clinical symptoms and MRI imaging findings of 35 patients obtained via a national survey.
Cerebral (type 1): Seizures, macrocephaly, and delayed psychomotor development are present, and cranial MRI characteristically shows cerebral white-matter lesions predominantly in the frontal region. It usually occurs in infancy or young childhood, and most cases are severe with a poor functional prognosis. In neonates it causes hydrocephalus and intracranial hypertension symptoms, and the survival prognosis is poor.
Bulbospinal (type 2): Hypotonia, spastic palsy, bulbar/pseudobulbar palsy, motor ataxia, and dysautonomia are present in a range of combinations, and MRI characteristically shows abnormalities or atrophy of the medulla oblongata and upper cervical spinal cord. It develops from childhood to adulthood and usually progresses more gradually than the other forms.
Intermediate (type 3): Possesses characteristics of both types 1 and 2. It occurs in a wide range of age groups, from infancy to adulthood.
In terms of age at onset and clinical signs, the infantile type corresponds closely to Prust et al.'s type 1 and Yoshida et al.'s type 1, and the clinical signs of the adult type correspond closely to Prust et al.'s type 2 and Yoshida et al.'s types 2 and 3. Patients with both cerebral and bulbospinal symptoms are classified as Yoshida et al.'s type 3, in the other two classifications, they may be allocated to any of the disease types; (Figure 1). In these guidelines, the classification used in the reference concerned is followed in the discussion of each subject.
Figure 1. Pathological classifications of Alexander disease and their relationships
2) Epidemiology
Frequency: A nationwide survey found that there are approximately 50 patients in Japan, a prevalence of one in 2.7 million people.4)
Sex difference: In the infantile type, boys tend to predominate, but no sex difference is present in the juvenile or adult forms.4)
Frequency of each type: Based on a GeneReview(R) report, the frequency of each type has been calculated as approximately 42% infantile, 22% juvenile, and 33% adult.7) The frequency of each type calculated from the Japanese nationwide survey was 27.3% infantile, 24.2% juvenile, and 48.5% adult.4)
Inheritance: Almost all infantile and juvenile cases are de novo mutations.4) Approximately 65% of adult cases occur in families, suggesting autosomal dominant inheritance.4) However, there have been few pedigree analyses, and its penetrance is unknown.
3) Etiology and pathophysiology
Transgenic mice engineered to express human wild-type GFAP exhibited shorter lifespans and the appearance of Rosenthal fibers in proportion to their GFAP expression levels,8) suggesting a gain-of-function mechanism. However, although ALXDRD patients carry GFAP mutations (the majority of which are missense mutations),2) transgenic mice with introduced mutant GFAP genes and knock-in model mice did not exhibit the full clinical phenotypes.9),10) This suggests that although a GFAP mutation is the necessary condition for the development of ALXDRD, upregulated GFAP expression may be a sufficient condition.
The following hypothesis has been suggested for the pathophysiology of ALXDRD.11)-13) Reduced proteasome function due to “mutant GFAP oligomers“ (coiled-coil polymers consisting of 4–6 molecules) formed as a result of GFAP mutations, causing mutant GFAP accumulation exceeding the toxic threshold and astrocyte activation in a positive feedback loop that further promotes mutant GFAP synthesis. Alongside the promotion of mutant GFAP accumulation, astrocyte function is impaired, including depletion of the molecular chaperone ab-crystallin, glutamate transporter abnormalities, and Rosenthal fiber formation.
However, the positive feedback loop involved in the promotion of GFAP synthesis, the toxic effect of Rosenthal fibers, and the details of astrocyte functional impairment have yet to been fully demonstrated.
No cases of human ALXDRD involving GFAP multiplication has been reported, suggesting the existence of modifying factors that affect quantitative changes in GFAP. GFAP promotor gene polymorphism has been reported as a candidate genetic modifying factor.14)
Findings of mouse model studies have suggested that demyelination, another pathological characteristic of ALXDRD, may be due to impaired myelin formation and maintenance as a result of dysfunctional potassium buffering,15) but the detailed mechanism is unknown.
4) Clinical symptoms
ALXDRD occurs in a wide range of age groups from neonates to individuals in their 70s.
Type 1: The three main symptoms are seizures, macrocephaly, and delayed psychomotor development.4 Seizures are typically intractable but lessen in some patients from later childhood. Macrocephaly is pronounced during infancy and childhood but less so during adulthood. Bulbospinal symptoms, including spastic palsy, dysarthria, dysphonia, and dysphagia, are progressive. Neonates are seriously affected, exhibiting hydrocephalus, intracranial hypertension, intractable seizures, and poor growth.
Type 2: Bulbospinal symptoms such as hypotonia of the limbs, spastic palsy, muscle rigidity, limb and trunk ataxia, dysarthria, dysphonia, dysphagia, dysautonomia (orthostatic hypotension, bladder and rectal disturbances, and sleep apnea), and cerebellar symptoms of varying severity are present.4) Motor symptoms usually exhibit laterality. Palatal tremor is uncommon, but in familial cases, it is strongly suggestive of ALXDRD. Seizures, macrocephaly, and delayed psychomotor development, the three main symptoms of type 1, are normally absent. A few cases of dementia have been reported, and some patients have exhibited cognitive dysfunction similar to that seen in frontotemporal dementia. One case of nodular lesions in the bilateral dorsal medulla oblongata has been reported in cases of MRI in which episodic vomiting was the only symptom.16),17)
Type 3: Characteristics of types 1 and 2 are present.4) Patients include those with type 1 who survive long-term and those with type 2 and delayed psychomotor development. The latter may not present on examination until type 2 symptoms become obvious in later childhood, and delayed psychomotor development may be identified at this point. Some patients previously suffered febrile convulsions or encephalopathy of unknown origin, and taking their medical history may reveal repeated seizures during infancy that improved or resolved in later childhood. Scoliosis and other spinal abnormalities and diplopia may also be present.
5) Investigations
5)-1 Imaging
Type 1: In a multicenter retrospective cranial MRI study of 217 patients with leukoencephalopathy, van der Knaap et al. reported that a high proportion of patients who met four or more of the following five criteria exhibited histopathological signs consistent with ALXDRD: (1) extensive cerebral white-matter changes with frontal predominance; (2) a periventricular rim with high signal intensity on T1-weighted images and low signal intensity on T2-weighted images; (3) abnormalities of the basal ganglia and thalamus; (4) brainstem abnormalities (particularly of the midbrain and medulla oblongata); and (5) contrast enhancement of at least one of the following: periventricular tissue, frontal white matter, optic chiasma, fornix, basal ganglia, thalamus, cerebellar dentate nucleus, or brainstem.5)
Type 2: Cranial MRI reveals atrophy and abnormal signal intensity of the medulla oblongata and cervical spinal cord in almost all cases.4) The brainstem typically exhibits a tadpole appearance, with the pons spared but severe atrophy of the medulla oblongata and upper cervical spine evident.18) In elderly individuals and mild cases with only slight bulbospinal atrophy, cranial MRI in the horizontal section may reveal an abnormal signal resembling an eye spot in the medullary pyramid.19) There are no abnormalities of the cerebrum, midbrain, or pontine pyramidal tract. In juvenile cases with onset from the late teens to twenties, abnormal nodular or tumor-like signals may be present in the medulla oblongata. A high proportion of patients also exhibit abnormal signals of the hilum of the cerebellar dentate nucleus20) and the midbrain periventricular rim on fluid-attenuated inversion recovery imaging.21)
Supratentorially, a ring-shaped hyperintensity described as a “periventricular garland” following the lateral ventricular wall is evident on T2-weighted imaging.6) This lesion may also exhibit contrast enhancement, and it is believed to be the location of large-scale Rosenthal fiber expression. Predominantly frontal cerebral white-matter lesions are absent, but a cap-shaped lesion may be present around the anterior horns of the lateral ventricles.
Type 3: Cerebral white matter lesions and bulbospinal atrophy and signal abnormalities, the characteristics of types 1 and 2, are both present.4) The severity of the cerebral white-matter lesions varies between patients; in many patients, they become cystic. Type 3 ALXDRD can be differentiated from megalencephalic leukoencephalopathy with subcortical cysts and vanishing white-matter disease by the fact that bulbospinal atrophy is always present.
5)-2 Genetic testing
A GFAP mutation is present in 98% of patients.2),7) Over 100 different types of GFAP mutation have been reported to date. The majority are missense mutations, but in-frame insertions and deletions, frame-shift mutations in the vicinity of the stop codon, and splice mutations have also been identified.22) Missense mutations are distributed throughout the GFAP domain but are more common in regions 1A, 2A, and 2B of the rod domain and the tail domain. The R79, R88, R239, and R416 positions in particular, which are associated with CpG, are hotspots for ALXDRD, with substitution mutations identified across different racial groups.2) R79, R88, and R239 substitution mutations are seen in types 1 and 3, and an R416 substitution mutation (R416W) has been reported in all three types.4) No mutation is particularly frequent in type 2.4)
6) Treatment and care
Treatment comprises only symptomatic therapy. Anti-epileptic drugs for seizures, nutritional management, antibiotics for comorbid infections, anti-spasmogenic drugs for spastic palsy, and special needs education and care for learning disorders and cognitive dysfunction are provided.7)
7) Diet and nutrition
Nutritional management, antibiotics for comorbid infections, and special needs education and care for learning disorders and cognitive dysfunction are provided.7) Motor dysfunction may include hypotonia, spastic palsy, and limb and trunk ataxia, but symptom severity and combination varies between individuals and rehabilitation should be based on an appropriate assessment. If dysphagia is present, it may be necessary to adjust the type of diet given, such as feeding thick purées.7)
8) Prognosis
The survival prognosis for type I is approximately 14 years.3) Neonates usually die of hydrocephalus and intracranial hypertension within a few weeks to a few months after birth. Many of those who develop the disease in infancy die of intractable seizures, malnutrition, infection, or other causes before they reach school age. However, in others, the seizures improve or resolve in later childhood, and despite the presence of intellectual disability, progressive gait disturbance, dysphagia, and other bulbospinal symptoms, these patients have a better prognosis than that of neonates or infants with intractable seizures. The survival prognosis for type II is approximately 25 years,3) better than that for type I, but there is wide individual variation in the functional prognosis of these patients, with some developing sudden aggravation of motor, bulbar, and respiratory symptoms23) and the symptoms remaining slight in others.24)
II.Diagnostic criteria
A. Neurological symptoms
- Seizures
- Macrocephaly
- Delayed psychomotor development
- Limb motor disability: hypotonia, spastic palsy, cerebral motor ataxia, muscle rigidity
- Bulbar or pseudobulbar palsy: dysphagia, dysarthria, dysphonia
- Dysautonomia: orthostatic hypotension, bladder and rectal disturbance, sleep apnea
- Palatal tremors 8. Recurrent vomiting
B. MRI findings
- Cerebral white-matter signal abnormalities with frontal predominance
- A periventricular rim that is hyperintense on T1-weighted imaging and hypointense on T2-weighted imaging
- Abnormalities of the basal ganglia and thalamus: hyperintense swelling or hyperintense/hypointense atrophy on T2-weighted imaging
- Contrast enhancement: periventricular tissue, frontal white matter, optic chiasma, fornix, basal ganglia, thalamus, cerebellar dentate nucleus, brainstem, or elsewhere
- Brainstem abnormalities or atrophy
1) Signal abnormalities of the midbrain
2) Medulla oblongata/upper cervical spine cord abnormalities
a) Spared pontine base with atrophy of the medulla oblongata and upper cervical spinal cord
b) Signal abnormalities of the medullary pyramid and cervical spinal cord on T2-weighted imaging
c) Non-atrophic nodular mass - Signal abnormalities or atrophy of the hilum of the cerebellar dentate nucleus
C. Genetic screening and pathological tests
- Genetic screening: identification of GFAP mutation
- Pathological tests: characteristic Rosenthal fibers present in the cytoplasm of cerebral white matter, subendymal, and subpial astrocytes
D. Differential diagnosis
Pelizaeus-Merzbacher disease and other congenital cerebral leukodystraphies as well as diseases with lesions mainly located in the cerebral white matter and medulla oblongata/spinal cord, including megalencephalic leukoencephalopathy with subcortical cysts, adrenoleukodystrophy, metachromatic leukodystrophy, merosin-deficient congenital muscular dystrophy, Krabbe disease, vanishing white-matter disease, Canavan disease, cerebrotendinous xanthomatosis, multiple sclerosis, neuromyelitis optica, acute disseminated encephalomyelitis, progressive multifocal leukoencephalopathy, brain tumor, cerebrovascular disturbance, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy, mitochondrial encephalomyopathy, hereditary spastic paraplegia, human T-lymphotropic virus-1–associated myelopathy, and amyotrophic lateral sclerosis.
Definitive diagnosis
Definite:
(1) At least one of A.1–3 and B.1
(2) At least one of A.4–8 and at least one of the findings in B.5.2)
Patients who exhibit either (1) or (2) above and either C.1 or C.2
Probable:
(1) At least one of A.1–3 or at least four of B.1–5
(2) At least one of A.4–8, at least one of the findings in B.5.2), and B.6
Patients who exhibit either (1) or (2) above and for whom the other conditions listed in the differential diagnosis (D) have been ruled out
Item (1) of the Definite and Probable criteria above corresponds to Yoshida et al.'s type 1 and item (2) to their type 2, with patients who exhibit both (1) and (2) corresponding to type 3.
III. Recent topics
Based on the hypothesized pathophysiology of ALXDRD, a number of studies have identified inhibited GFAP expression and the upregulation of ab-crystallin expression as potential targets for therapeutic drugs. Experiments at the cellular level have been reported for the third-generation cephem antibiotic ceftriaxone25) and curcumin, a polyphenol.26) The antipsychotic lithium has been reported as effective in a murine model.27) However, its narrow therapeutic window and high toxicity render its use problematic.27) Other potential therapeutic strategies include studies implicating glial-derived nitric oxide (NO) in non-cell autonomous neurodegeneration28) and the inhibition of oxidative stress by muscarinic acetylcholine receptor antagonists.29) The generation of induced pluripotent stem cells from the skin or blood of ALXDRD patients has also been reported,30) which may provide an important tool for studying its pathophysiology and treatment.
References
(Unless otherwise noted at the end, all are evidence level VI.)
- Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 1949: 72: 373-381.(V)
- Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 2001: 27: 117-120.(IVb)
- Prust M, Wang J, Morizono H, Messing A, Brenner M, Gordon E, Hartka T, Sokohl A, Schiffmann R, Gordish-Dressman H, Albin R, Amartino H, Brockman K, Dinopoulos A, Dotti MT, Fain D, Fernandez R, Ferreira J, Fleming J, Gill D, Griebel M, Heilstedt H, Kaplan P, Lewis D, Nakagawa M, Pedersen R, Reddy A, Sawaishi Y, Schneider M, Sherr E, Takiyama Y, Wakabayashi K, Gorospe JR, Vanderver A. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology 2011: 77: 1287-1294.(IVb)
- Yoshida T, Sasaki M, Yoshida M, Namekawa M, Okamoto Y, Tsujino S, Sasayama H, Mizuta I, Nakagawa M. Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J Neurol 2011; 258: 1998-2008. (IVb)
- van der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S, Begger JC, van Coster R, Barth PG, Thomas NH, Valk J, Powers JM. Alexander disease: diagnosis with MR imaging. Am J Neuroradiol 2001: 22: 541-552.(IVb)
- van der Knaap MS, Ramesh V, Schiffmann R, Blaser S, Kyllerman M, Gholkar A, Ellison DW, van der Voorn JP, van Dooren SJM, Jakobs C, Barkhof F, Salomons GS. Alexander disease. Ventricular garlands and abnormalities of the medulla and spinal cord. Neurology 2006: 66: 494-498.(V)
- Srivastava S,Naidu S. Alexander disease. GeneReviews(R)[Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. 2002 Nov 15 [updated 2015 Jan 8].
- Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol 1998: 152: 391-398.
- Hagemann TL, Connor JX, Messing A. Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and white matter stress response. J Neurosci 2006: 26: 11162-11173.
- Tanaka FK, Takebayashi H, Yamazaki Y, Ono K, Naruse M, Iwasato T, Itohara S, Kato H, Ikenaka K. Murine model of Alexander disease: analysis of GFAP aggregate formation and its pathological significance. GLIA 2007: 55: 617-631.
- Tang G, Xu Z, Goldman JE. Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem 2006: 281: 38634-38643.
- Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE. Oligomers of mutant glial fibrillary acidic protein (GFAP) inhibit the proteasome system in Alexander disease astrocytes, and the small heat shock protein αB-crystallin reverses the inhibition. J Biol Chem 2010: 285: 10527-10537.
- Hagemann TL, Boelens WC, Wawrousek EF, Messing A. Suppression of GFAP toxicity by B-crystallin in mouse models of Alexander disease. Hum Mol Genet 2009: 18: 1190-1199.
- Yoshida T, Mizuta I, Saito K, Ohara R, Kurisaki H, Ohnari K, Riku Y, Hayashi Y, Suzuki H, Shii H, Fujiwara Y, Yonezu T, Nagaishi A, Nakagawa M. Effects of a polymorphism in the GFAP promoter on the age of onset and ambulatory disability in late-onset Alexander disease. J Hum Genet 2013: 58: 635-638.(V)
- Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE. Alexander disease. J Neurosci 2012: 32: 5017-5023.
- Niinikoski H, Haataja L, Brander A, Valanne L, Blaser S. Alexander disease as a cause of nocturnal vomiting in a 7-year-old girl. Pediatr Radiol 2009: 39: 872-875.(V)
- Namekawa M, Takiyama Y, Honda J, Sakoe K, Naoi T, Shimazaki H, Yamagata T, Momoi MY, Nakano I. A novel adult case of juvenile-onset Alexander disease: complete remission of neurological symptoms for over 12 years, despite insidiously progressive cervicomedullary atrophy. Neurol Sci 2012: 33: 1389-1392.(V)
- Namekawa M, Takiyama Y, Honda J, Shimazaki H, Sakoe K, Nakano I. Adult-onset Alexander disease with typical “tadpole” brainstem atrophy and unusual bilateral basal ganglia involvement: a case report and review of the literature. BMC Neurol 2010: 10: 21.(V)
- Yoshida T, Mizuta I, Saito K, Kimura Y, Park K, Ito Y, Haji S, Nakagawa M, Mizuno T. Characteristic abnormal signals in medulla oblongata-‘eye spot’ sign: four cases of elderly-onset Alexander disease. Neurol Clin Pract 2015: 5: 259-262.(V)
- Graff-Radford J, Schwartz K, Gavrilova RH, Lachance DH, Kumar N. Neuroimaging and clinical features in type II (late-onset) Alexander disease. Neurology 2014; 82: 49-56.(V)
- Farina L, Pareyson D, Minati L, Ceccherini I, Chiapparini L, Romano S, Gambaro P, Fancellu R, Savoiardo M. Can MR imaging diagnose adult-onset Alexander disease? Am J Neuroradiol 2008: 29: 1190-1196.(V)
- Flint D, Li R, Webster LS, Naidu S, Kolodny E, Percy A, van der Knaap M, Powers JM, Mantovani JF, Ekstein J, Goldman JE, Messing A, Brenner M. Splice site, frameshift, and chimeric GFAP mutations in Alexander disease. Hum Mutat 2012; 33: 1141-1148.(V)
- Ayaki T, Shinohara M, Tatsumi S, Namekawa M, Yamamoto T. A case of sporadic adult Alexander disease presenting with acute onset, remission and relapse. J Neurol Neurosurg Psych 2010: 81: 1292-1293.(V)
- Sugiyama A, Sawai S, Ito S, Mukai H, Beppu M, Yoshida T, Kuwabara S. Incidental diagnosis of an asymptomatic adult-onset Alexander disease by brain magnetic resonance imaging for preoperative evaluation. J Neurol Sci 2015; 354: 131-132.(V)
- Bachetti T, Zanni ED, Balbi P, Bocca P, Prigione I, Deiana GA, Rezzani A, Ceccherini I, Sechi GP. In vitro treatments with ceftriaxone promote elimination of mutant glial fibrillary acidic protein and transcription down-regulation. Exp Cell Res 2010: 316: 2152-2165.
- Bachetti T, Di Zanni E, Balbi P, Ravazzolo R, Sechi GP, Ceccherini I. Beneficial effects of curcumin on GFAP filament organization and down-regulation of GFAP expression in an in vitro model of Alexander disease. Exp Cell Res 2012: 318: 1844-1854.
- LaPash Daniels CM, Paffenroth E, Austin EV, Glebov K, Lewis D, Walter J, Messing A. Lithium decreases glial fibrillary acidic protein in a mouse model of Alexander disease. PLoS One 2015: 10: e0138132.
- Wang L, Hagemann TL, Kalwa H, Michel T, Messing A, Feany MB. Nitric oxide mediates glial-induced neurodegeneration in Alexander disease. Nat Commun 2015; 6: 8966.
- Wang L, Hagemann TL, Messing A, Feany MB. An in vivo pharmacological screen identifies cholinergic signalling as a therapeutic target in glial-based nervous system disease. J Neurosci 2016; 36: 1445-1455.
- Kondo T, Funayama M, Miyake M, Tsukita K, Era T, Osaka H, Ayaki T, Takahashi R, Inoue H. Modeling Alexander disease with patient iPSCs reveals cellular and molecular pathology of astrocytes. Acta Neuropathol Comm 2016; 4: 69.
Literature search
1. Overview
1)Definition
- PubMed (search date 2016/12/27)
“Alexander Disease”[MeSH Major Topic] 195 results - Igaku Chūō Zasshi ( search date 2016年12月27日)
((Alexander病/TH or アレキサンダー病/AL)) and (PT=会議録除く) 88 results
2)Epidemiology
- PubMed (search date 2016/12/27)
“Alexander Disease/epidemiology”[Mesh] 4 results - Igaku Chūō Zasshi (search date 2016/12/27)
((Alexander病/TH or アレキサンダー病/AL)) and ((有病率/TH or 有病/AL))and (PT=会議録除く) 1 results
3)Etiology and pathophysiology
- PubMed (search date 2016/12/27)
“Alexander Disease/etiology”[Majr] 106 results - Igaku Chūō Zasshi (search date 2016/12/27)
((病態生理/TH or 病態/AL))or病因/AL)and ((Alexander病/TH or アレキサンダー病/AL)) and (PT=会議録除く) 21 results - Hand research
4)Clinical symptoms
- PubMed (search date 2016/12/27)
“Alexander Disease”[Majr] AND “humans”[MeSH Terms] 184 results - Igaku Chūō Zasshi (search date 2016/12/27)
((Alexander病/TH or アレキサンダー病/AL)) and ((徴候と症状/TH or 症状/AL)) and (PT=会議録除く) 17 results
5)Investigations
5)-1 Imaging
5)-2 Genetic testing
6) Treatment and care
7)Diet and nutrition
- PubMed (search date 2016/12/27)
“Alexander Disease”[MeSH Major Topic] 195 results - Igaku Chūō Zasshi (search date 2016/12/27)
((Alexander病/TH or アレキサンダー病/AL)) and (PT=会議録除く) 88 results
8)Prognosis
- PubMed (search date 2016/12/27)
((“prognosis”) OR “mortality”) AND “Alexander Disease”[MeSH Major Topic] 5 results - Igaku Chūō Zasshi (search date 2016/12/27)
((Alexander病/TH or アレキサンダー病/AL)) and ((予後/TH or 予後/AL))and (PT=会議録除く) 2 results
3. Recent topics
- PubMed (search date 2016/12/27)
“Alexander Disease”[Majr] AND (“2014/01/01”[PDAT] : “2016/12/31”[PDAT]) 28 results - Igaku Chūō Zasshi (search date 2016/12/27)
((Alexander病/TH or アレキサンダー病/AL)) and (DT=2011:2016 PT=会議録除く) 24 results