








Diagnostic imaging of congenital cerebral hypomyelination
Junichi TAKANASHI, Department of Pediatrics, Tokyo Women's Medical University Yachiyo Medical Center
Cerebral hypomyelination includes a group of diseases characterized by impaired or delayed myelin formation. Imaging findings for these diseases resemble those of neonates with immature myelination (i.e., compared to the cortex, the white matter appears hypointense on T1-weighted images and hyperintense on T2-weighted images). T2-weighted images show that the entire white matter is faintly hyperintense, with only those sites where early myelination occurs (such as the posterior limb of the internal capsule, the optic radiation and the corticospinal tract) exhibiting hypointensity reflecting myelination.1-3) Based on the changes in magnetic resonance (MR) intensity associated with myelination, T1 hyperintensity is known to develop before T2 hypointensity. The appearance of T1-weighted images varies depending on the degree of myelination, ranging from overall, faint hypointensity of the white matter, to faint hyperintensity seen only in areas where myelination occurs at an early stage, and finally to faint hyperintensity over a wide area. A definitive diagnosis of hypomyelination can be made on the basis of magnetic resonance imaging (MRI) findings if the abnormal white matter signal intensity is unchanged over a period of at least 6 months, with at least one MRI evaluation carried out after age 1. If severe hypomyelination is apparent on MRI after age 2, cerebral hypomyelination is highly likely.2, 3) The imaging findings for the various diseases classified as cerebral hypomyelination are described below (Table 1).
Table 1. MRI findings for congenital cerebral hypomyelination
| Disease name | OMIM | Gene | Type of inheritance | Peripheral neuropathy | Clinical presentation | Imaging findings |
|---|---|---|---|---|---|---|
| Pelizaeus-Merzbacher disease (PMD) | HLD1, 312080 | PLP1 | X-linked | Congenital nystagmus, head tremors, spastic paralysis, involuntary movements | Widespread T1 hypointensity, T2 hyperintensity [hypomyelination pattern (HP)], high NAA on MRS | |
| Pelizaeus-Merzbacher like disease (PMLD) | HLD2, 608804 | GJC2 | AR | Congenital nystagmus, head tremors, spastic paralysis, involuntary movements | HP, corticospinal tract comparatively preserved. Hyperintensities in the corticospinal tract on difusion-weighted imaging (reduced diffusion) | |
| Hypomyelinating leukodystrophy-3 | HLD3, 260600 | AIMP1 | AR | Microcephaly, nystagmus, severe mental disability, motor developmental retardation | HP, cerebral atrophy, thinning of the corpus callosum | |
| HSP60 chaperonopathy | HLD4, 612233 | Hsp60 (HSPD1) | AR | Nystagmus, spasticity, developmental retardation and regression, epilepsy | HP, thinning of the corpus callosum and ventricular enlargement, atrophy of the brainstem and cerebellum, T2 hyperintensities in the brainstem. Hyperintensities in the corticospinal tract on diffusion-weighted imaging (decreased ADC) | |
| Hypomyelination and congenital cataract (HCC) | HLD5, 610532 | FAM126A | AR | (+) | Congenital cataract, mental disability, motor developmental retardation, peripheral neuropathy (decreased tendon reflexes) | HP, strong T2 hyperintensities and T1 hypointensities in the periventricular and deep white matter, with progressive atrophy in these regions over time |
| Hypomyelination with atrophy of the basal ganglia and cerebellum (HABC) | HLD6,612438 | TUBB4A | AR | Mental disability, motor develomental retardation, spasticity, cerebellar ataxia, extrapyramidal tract symptoms (athetosis, dystonia, and rigidity) | HM, progressive atrophy of white matter in the cerebellum (vermiform process > hemispheres), head of the caudate nucleus and putamen, and cerebrum. Some patients may not exhibit atrophy of the basal ganglia (+ cerebellum). | |
| Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome) | HLD7, 607694, HLD8, 614381, HLD11, 616494 | POLR3A, POLR3B, POLR1C | AD | (±) | Mental disability, progressive ataxia, oligodontia/adontia and other forms of hypodontia, hypogonadotropic hypogonadism | HM, cerebellar atrophy and thinning of the corpus callosum |
| Hypomyelinating leukodystrophy-9 | HLD9, 616140 | RARS | AR | Motor developmental retardation, mental disability, spasticity, nystagmus | HM, thinning of the corpus callosum | |
| Hypomyelinating leukodystrophy-10 | HLD10, 616420 | PYCR | AR | Microcephaly, motor develomental retardation, mental disability | HM, decreased cerebellar and white matter volume, thinning of the corpus callosum, brainstem hypoplasia | |
| 18q-deletion syndrome | 601808 | 18q- (MBP?) | AD | Short stature, mental disability, motor develomental retardation, seizures | Diffuse hyperintensity may be evident on T2-weighted imaging, but patchy hyperintensity is often present. | |
| Allan-Herndon-Dudley syndrome (AHDS) | 300523 | SLC16A2 (MCT8) | X-linked | Mental retardation, dysarthria, athetoid movement, hypotonia, spastic paraplegia | HM, with progressive myelination over time | |
| Salla disease | 604369 | SLC17A5 | AR | Developmental retardation, developmental regression, poor physical growth, ataxia,nystagmus, hypotonia, spasticity, epilepsy | HM, high NAA (NANA) on MRS | |
| Peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease (PCWH) |
609136 | SOX10 | AD, sporadic | (+) | Hirschsprung disease, Waardenburg syndrome, motor developmental retardation, mental disability, peripheral neuropathy | HM, cerebral atrophy, delayed myelination in mild cases |
| Oculodentodigital dysplasia (ODDD) | 164200 | GJA1 | AD | Eyes (small eyes), teeth (enamel hypoplasia), fingers (syndactylia of the 4th and 5th fingers) | HM, T2 hypointensity of the globus pallidus, cerebellar atrophy | |
| Trichothiodystrophy with hypersensitivity to sunlight (Tay syndrome) | 601675 | ERCC2, ERCC3, GTF2H5, MPLKIP | AR | Motor develomental retardation, mental disability, impaired growth, coarse and brittle hair, ichthyosiform skin | HM, cerebellar atrophy | |
| Hypomyelination with brain stem and spinal cord involvement and leg spasticity (HBSL) | 615281 | DARS | AR | Motor developmental retardation, mental disability, nystagmus, spasticity (arms < legs) | HM (of the cerebellum, brainstem, and dorsal funiculus as well as the cerebral white matter), thinning of the corpus callosum | |
| Fucosidosis | 230000 | FUCA1 | AR | Motor developmental retardation, mental disability, facial abnormalities, bone deformities, elevated NaCl concentration in sweat, hepatosplenomegaly, cardiac hypertrophy, seizures, susceptibility to infection. Mild form (type 2) mainly angiokeratoma | HM, T2 hypointensity and T1 hyperintensity of the globus pallidus, T2 hypointensity of the thalamus, cerebellar atrophy |
1) Pelizaeus-Merzbacher disease (PMD, HLD1, OMIM#312080)
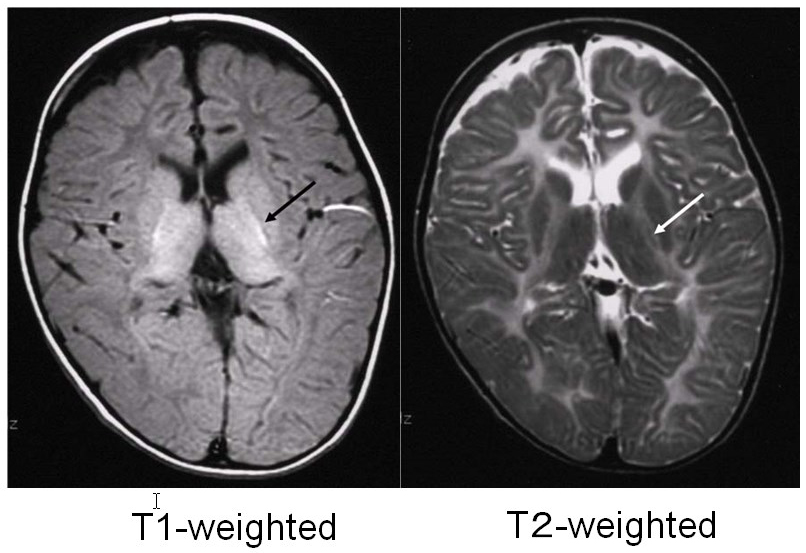
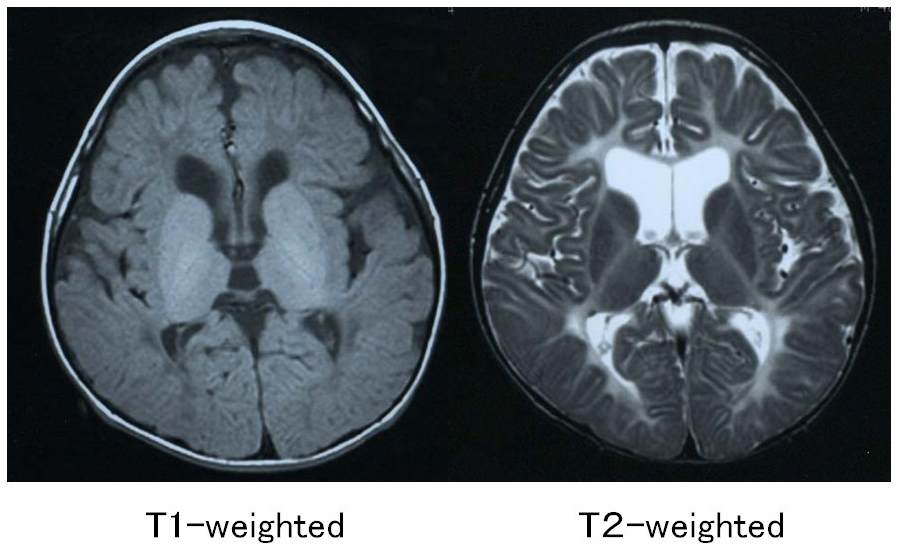
Pelizaeus-Merzbacher disease is an X-linked recessive disorder caused by a defect in the proteolipid protein 1 (PLP1) gene, which encodes the main constituent protein of myelin. Its symptoms include congenital nystagmus, head tremors, spastic paralysis, and involuntary movements. It is broadly classified into a congenital type that appears soon after birth and a classical type that develops after several months. In 70% of affected individuals, the PLP1 gene is duplicated, and most of these individuals develop the classical type of the disorder. MRI findings are correlated with clinical signs: individuals with the congenital type of the disorder exhibit no myelination at all, with widespread T1 hypointensity and T2 hyperintensity of the white matter (Figure 1), whereas in individuals with the classical type, myelination is present in locations including the posterior limb of the internal capsule, the optic radiation and the corticospinal tract, all of which appear hyperintense on T1-weighed images and hypointense on T2-weighted images (Figure 2).4)
2) Pelizaeus-Merzbacher-like disease (PMLD, HDL2, OMIM#608804)
When individuals exhibit symptoms that are clinically indistinguishable from those of PMD but do not possess a PLP1 gene defect, this is referred to as Pelizaeus-Merzbacher-like disease (PMLD) in order to distinguish it from PMD. Some affected individuals exhibit a GJC2 gene defect and are diagnosed with PMLD1. The condition exhibits autosomal recessive inheritance and may also occur in females. Nystagmus is evident soon after birth, and motor developmental retardation is noticeable before the age of 1 year. Pyramidal tract, cerebellar, and basal ganglia symptoms subsequently appear. MRI findings resemble those of PMD, in that diffuse white matter signal abnormalities are present, but the corticospinal tract is comparatively unaffected (T2 hypointensity).5, 6) The corticospinal tract may appear hyperintense (reduced diffusion) on diffusion-weighted imaging.7)
3) Hypomyelinating leukodystrophy-3 (HLD3, OMOM#260600)
This form of cerebral hypomyelination is caused by a defect in the AIMP1 gene, and its symptoms include microcephaly, nystagmus, severe mental disability and motor developmental retardation. Some authorities consider that it should be regarded as a form of cortical degeneration because of the presence of microcephaly and brainwave abnormalities. No case has been reported from Japan. MRI reveals white matter that appears hyperintense on T2-weighted imaging and faintly hypointense on T1-weighted imaging, consistent with hypomyelination.8) Cerebral atrophy and thinning of the corpus callosum are present. It has been reported that magnetic resonance spectroscopy (MRS) reveals relatively low N-acetylaspartate (NAA) levels.8)
4) HSP60 chaperonopathy (HLD4, OMIM#612233)
This form of congenital cerebral hypomyelination is caused by a defect in the mitochondrial heat-shock protein 60 (Hsp60). The gene associated with this disorder is the Hsp60 gene (HSPD1), and no case has been reported from Japan. Nystagmus, spasticity, developmental retardation and regression are present, and epilepsy may occur in approximately half of the affected individuals. The cerebral white matter exhibits diffuse T2 hyperintensity, with no myelination at all. 9) Thinning of the corpus callosum, ventricular enlargement and atrophy of the brainstem and cerebellum are evident. The brainstem appears hyperintense on T2-weighted imaging, and the corticospinal tract is hyperintense on diffusion-weighted imaging (decreased ADC). Reports have stated that MRS reveals high myo-inositole (mIns)/creatine (Cr), normal choline (Cho)/Cr, and normal or low NAA/Cr.9)
5). Hypomyelination and congenital cataract (HCC, HLD5, OMIM#610532)
This is an autosomal recessive condition characterized by cerebral hypomyelination and congenital cataract, caused by a defect of the FAM126A gene. MRI reveals diffuse T2 hyperintensity consistent with hypomyelination (with the posterior limb of the internal capsule also hyperintense).10, 11) The periventricular and deep cortex is more strongly hyperintense on T2-weighted imaging and hypointense on T1-weighted imaging, and atrophy at these sites progresses over time.11) Studies have suggested that the water content of the white matter may be higher in affected individuals. White matter damage (demyelination) other than hypomyelination must also be considered.
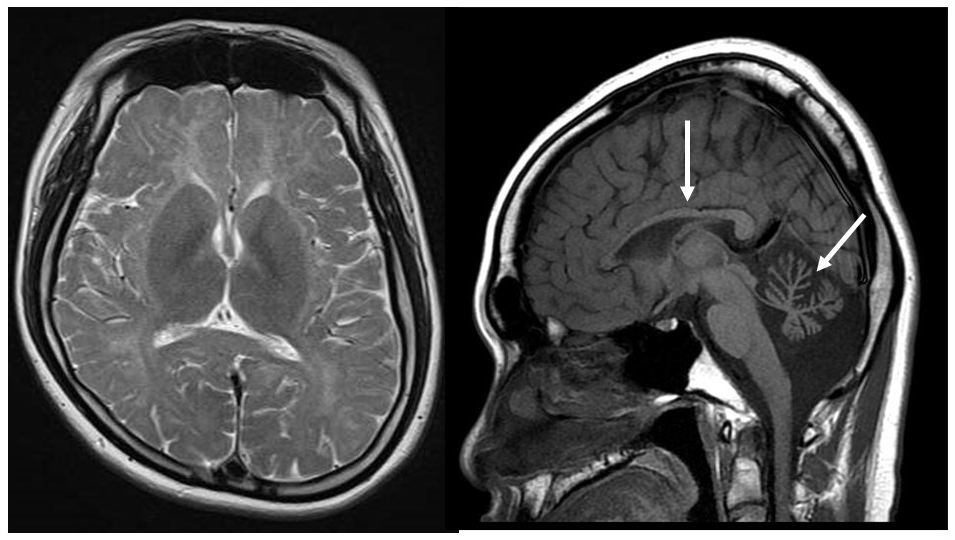
6) Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC, HLD6).
This is a new form of white matter degeneration caused by a TUBB4A gene defect, which results in the delay and subsequent regression of motor development, extrapyramidal tract symptoms (dystonia), ataxia, and spastic paralysis. Mental development is comparatively unaffected. Slight hypointensities may be evident in the corpus callosum and the posterior limb of the internal capsule on T2-weighted imaging, but they are almost entirely hyperintense.12, 13) Their appearance on T1-weighted imaging varies from hypointense to slightly hyperintense. Progressive atrophy of the cerebellum (vermiform process > hemispheres), the head of the caudate nuclei and the putamen, and the cerebral white matter are also apparent (Figure 3). The globus pallidus and the thalamus are preserved. Some patients with TUBB4A gene defects exhibit cerebellar atrophy without any atrophy of the basal ganglia, which makes HDL6 difficult to distinguish from HDL7 and HDL8 on the basis of imaging findings alone.13)
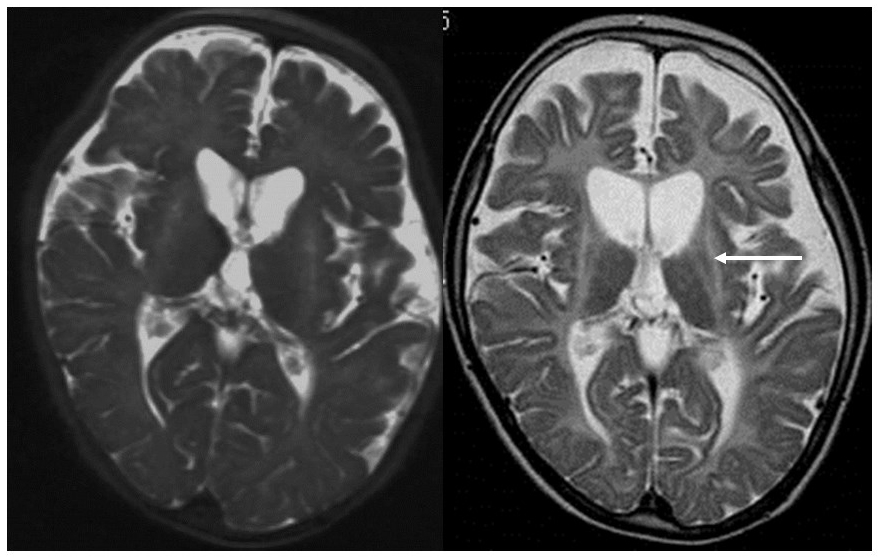
T2-weighted imaging during infancy (left) revealed hyperintensity of the white matter and atrophy of the basal ganglia and the cerebellum. T2-weighted imaging at elementary school age (right) revealed that the atrophy of the basal ganglia (arrow) and the cerebellum had progressed.
7) Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome, HLD7, OMIM#607694, HLD8, OMIM#614381, HLD11, OMIM#616494)
These forms of white matter degeneration are caused by defects in the POLR3A (HLD7) and POLR3B (HLD8) genes, and are characterized by mental disability, progressive ataxia, oligodontia, adontia, and other forms of hypodontia, and hypogonadotropic hypogonadism. T2-weighted MRI imaging reveals diffuse hyperintensity of the cerebral white matter.14, 15) Cerebellar atrophy and thinning of the corpus callosum are also present, and this disorder corresponds to a group of affected individuals from Japan reported to be suffering from hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum (HCAHC) (Figure 4).14) POLR3B (HLD8) defects cause more severe cerebellar atrophy than do those in PLOR3A (HLD7), and it is believed that myelination has progressed.16) A recent study has also found that POLR1C gene defects also cause similar clinical symptoms and imaging findings.17)
A man in his 20s exhibiting developmental retardation and hypogonadism with a POLR3B gene defect. T2-weighted imaging revealed hyperintensity of the cerebral white matter with cerebellar atrophy and hypoplasia of the corpus callosum (arrows).
8) Hypomyelinating leukodystrophy-9 (HLD9, OMOM#616140)
This autosomal recessive condition is caused by a RARS gene defect, and causes motor developmental retardation, mental disability, spasticity, and nystagmus during infancy. Imaging findings are consistent with hypomyelination, with diffuse T2 hyperintensity of the cerebral white matter (including the internal capsule).18) MRS has been reported to show low Choline.
9) Hypomyelinating leukodystrophy-10 (HLD10, OMOM#616420)
This autosomal recessive condition is caused by a PYCR gene defect, and causes microcephaly, motor developmental retardation, and mental disability. The white matter is hyperintense on T2-weighted imaging, but the corpus callosum and the internal capsule are hypointense. Microcephaly, reduced white matter volume, thinning of the corpus callosum, and brainstem hypoplasia are also present.19) It is also regarded as a category of cortical degeneration on the basis of its clinical presentation and imaging findings.
10) 18q deletion syndrome (601808)
Affected individuals exhibit short stature, psychomotor retardation, hypotonia and seizures. Dysplasia of the head and face is evident, and deformities such as microcephaly, flattened facial features, orbital hypertelorism, heart malformations, syndactyly, deafness, and cleft palate are also common. The prognosis for survival is good. The cerebral white matter may be diffusely hyperintense on T2-weighted imaging, but patches of hyperintensity are frequently present. The corpus callosum and the posterior limb of the internal capsule are often hypointense on T2-weighted imaging. The cerebral white matter is isiontense or faintly hyperintense on T1-weighted imaging.20) These imaging findings are not very suggestive of hypomyelination. It has been believed that hypomyelination is caused by the loss of the basic myelin protein gene (18q22-23), although some studies have found that pathological tests reveal normal myelin, with the T2 hyperintensity actually due to astrogliosis.20, 21)
11) Allan-Herndon-Dudley syndrome (AHDS, OMIM#300523)
This is a type of X-linked mental retardation syndrome. Its main symptoms include severe mental retardation, dysarthria, athetoid movement, hypotonia, and spastic paraplegia; MRI reveals that myelination is delayed at infancy. The cerebral white matter exhibits diffuse hyperintensity on T2-weighted imaging, but the corpus callosum and the posterior limb of the internal capsule often exhibit T2 hypointensity.22) On T1-weighted imaging, it appears faintly hyperintense. In most cases myelination progresses over time, and strictly speaking, it is considered to constitute delayed myelination. MRS has been reported to show high Cho and mIns.22)
12) Salla disease (OMIM#604369)
This is an autosomal recessive disease characterized by the accumulation of sialic acid in lysosomes. The causative gene is SLC17A5 (6q14-q15). The same gene mutation causes infantile sialic acid storage disorder (ISSD, OMIM#269920), which is fatal in infancy and a slow-progressing, mild disorder during adulthood. The cerebral white matter exhibits diffuse hyperintensity on T2-weighted imaging, but the posterior limb of the internal capsule and the radiate crown may display T2 hypointensity (T1 hyperintensity).23) Thinning of the corpus callosum is evident. MRS reveals high NAA, but it is believed that this is because MRS is incapable of distinguishing between N-acethylneuraminic acid (NANA) and NAA.
13) Peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease (PCWH, OMIM#609136)
This is a rare disease characterized by the dysgenesis not only of oligodendrocytes, but also of Schwann cells, melanocytes, intestinal ganglion cells and other neural crest-derived cells. Clinically, it combines four different syndromes, including hypomyelination of the central nervous system and demyelinating neuropathy in the peripheral nervous system, in addition to Waardenburg syndrome and Hirschsprung disease. The gene associated with this disorder is the autosomal dominant SOX10 gene, with most cases being sporadic due to a sudden mutation. The entire cortical white matter exhibits diffuse hyperintensity on T2-weighted imaging, and cerebral atrophy is present.24) Mild cases, however, exhibit signs indicative of delayed myelination, with myelination occurring gradually over time.25)
14) Others
Other disorders that may exhibit a hypomyelination pattern on diagnostic imaging include Hypomyelination with brain stem and spinal cord involvement and leg spasticity (HBSL, DARS gene defect), Trichothiodystrophy with hypersensitivity to sunlight (Tay syndrome), Fucosidosis, Serine synthesis defects, Oculodentodigital dysplasia (ODDD), Galactosemia, Cockayne syndrome, early onset neuronal degenerative disorders (early onset GM1, GM2 gangliosidosis, infantile neuronal ceroid lipofuscinosis, Alpers syndrome).
References
- Takanashi J. Hakushitsu henseishou no gazou shindan [Diagnostic imaging of degenerative white matter disorders]. Journal of the Japan Pediatric Society. 2007; 111: 1243-1254. [In Japanese]
- Schiffmann R, van der Knaap MS. An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009; 72: 750-759
- Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010; 133; 2971-2982.
- Takanashi J, Sugita K, Tanabe Y, et al. MR-revealed myelination in the cerebral corticospinal tract as a marker for Pelizaeus-Merzbacher's disease with proteolipid protein gene duplication. AJNR Am J Neuroradiol 1999; 20: 1822-1828.
- Uhlenberg B, Schuelke M, Ruschendorf F, et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004; 75:251–260.
- Abrams CK, Scherer SS. Gap junctions in inherited human disorders of the central nervous system. Biochem Biophys Acta 2012; 1818; 2030-2047.
- Biancheri R, Rosano C, Denegri L, et al. Expanded spectrum of Pelizaeus–Merzbacher-like disease: literature revision and description of a novel GJC2 mutation in an unusually severe form. Eur J Hum Genet 2013; 21: 34-39.
- Feinstein M, Markus B, Noyman I, et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am J Hum Genet 2010; 87: 820-828.
- Magen D, Georgopoulos C, Bross P, et al. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet 2008; 83: 30-42.
- Zara F, Biancheri R, Bruno C, et al. Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat Genet 2006; 38: 1111-3.
- Biancheri R, Zara F, Rossi A, et al. Hypomyelination and congenital cataract. Broadening the clinical phenotype. Arch neurol 2011; 68: 1191-1194.
- Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 2013; 92: 767-73.
- Miyatake S, Osaka H, Shiina M, et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology 2014; 82: 2230-7.
- Sasaki M, Takanashi J, Tada H, et al: Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev 2009; 31: 582-587.
- Saitsu H, Osaka H,Sasaki M, et al: Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal recessive hypomyelinating leukoencephalopathy. Am J Hum Genet 2011; 89: 644-651.
- Takanashi J, Osaka H, Saitsu H, et al. Different patterns of hypomyelination and cerebellar abnormality between POLR3A and POLR3B mutations. Brain Dev 2014; 36: 259-263.
- Thiffault I, Wolf NI, Forget D, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun in press. doi: 10.1038/ncomms8623
- Wolf NI, Salomons GS, Rodenburg RJ, et al. Mutations in RARS cause hypomyelination. Ann Neurol 2014; 76: 134-139.
- Nakayama T, Al-Maawali A, El-Quessny M, et al. Mutations in PYCR2, encoding pyrroline-5-carboxylate reductase 2, cause microcephaly and hypomyelination. Am J Hum genet 2015 96: 709-719.
- Tada H, Takanashi J. MR spectroscopy in 18q- syndrome suggesting other than hypomyelination. Brain Dev 2014; 36: 57-60.
- Tanaka R, Iwasaki N, Hayashi M, et al. Abnormal brain MRI signal in 18q- syndrome not due to dysmyelination. Brain Dev 2012; 34: 234–237.
- Gika AD, Siddiqui A, Hulse AJ, White matter abnormalities and dystonic motor disorder associated with mutations in the SLC16A2 gene. Dev Med Child Neurol 2010; 52: 475-482.
- Sonninen P, Autti T, Varho T, et al. Brain involvement in Salla disease. Am J Neuroradiol 1999; 20: 433-443.
- Inoue K, Tanabe Y, Lupski JR. Myelin deficiencies in both the central and the peripheral nervous system associated with a SOX10 mutation. Ann Neurol 1999; 46: 313-318.
- Verheij JB, Sival DA, van der Hoeven JH, et al. Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: a case report and review of the literature. Eur J Paediatr Neurol. 2006; 10:11-17.