




遺伝性白質疾患ガイドライン
ペリツェウス・メルツバッハ病 Pelizaeus-Merzbacher disease(PMD、HLD1、OMIM#312080)
疾患説明; プロテオリピドプロテイン1(proteolipid protein 1; PLP1)遺伝子の異常による中枢神経系の髄鞘形成不全症、X連鎖性劣性遺伝形式をとるため、患者は例外を除き男児のみである。先天性眼振を呈し、乳児には低緊張を呈し、その後形成、固縮、小脳失調が明らかとなる。重症例では、頸定が得られないが独歩を獲得する軽症例もある。運動発達に比し、知的発達は良好である。
治療; 臍帯血移植、幹細胞移植が行われているが、少数例であり劇的な改善は得られていない。現時点では対症的な治療が行われる。
1. 概要
定義
Friedrich Pelizaeus(1885,ドイツ人医師)Ludwig Merzbacher(1910,ドイツ人病理学者)により最初に記述され、後にSeitelberger(1970)により確立された病理・臨床学的な症候群(1)。男児に見られる、中枢神経の髄鞘形成不全症であり病理学的には、“白質のミエリン鞘が消失・あるいは極度に低下するがニューロンは保持され、あきらかな脱髄の変化を伴わず、残存したミエリンが斑状のtigroidの像を呈する”ことを特徴とする。その後の遺伝学的な解析により、プロテオリピドプロテイン1(proteolipid protein 1; PLP1)の遺伝子異常がこの疾患の主たる原因であることが明らかにされた(2, 3)。PLP1がミエリン蛋白の主要な分子であり、中枢神経のオリゴデンドロサイトに発現するため、中枢神経系に限局した症状を示すミエリン形成不全症である(4)。
疫学
本邦での発症率は、10万人出生あたり、0.26人とされ、アメリカ、ドイツからの報告と類似している(5)。
病因・病態、
中枢神経系のアクソン周囲を電気的に絶縁するミエリンは、約80%の脂質と20%の蛋白質とからなる。その蛋白質中約50%を占め、最も量的に豊富なのが、Proteolipid protein1 (PLP1)である。PLP1遺伝子は、7つのエクソンから成り、PLP1とDM 20の2つの蛋白質をコードし、いずれも4回膜貫通型の構造をとると考えられている(図1 A, B)(3)。DM 20はPLP1の一部(エクソン3の後半部分に相当する35アミノ酸)を欠いており、胎生期はPLP1より優位に、種々の臓器に発現し、下等生物ではDM 20の相同分子のみ認める。PLP1は出生後にその発現が上昇し、中枢オリゴデンドロサイトに限局した発現をみる。このPLP1遺伝子を欠損したマウスでは、ミエリン鞘のコンパクトな層状構造が失われ、ミエリン層のスペースが開大し、中枢伝導時間が延長することが確かめられており、PLP1はミエリンの膜接合と安定化に関係すると考えられている(図1C)(5)。またこのマウスのみならず欠損した患者においてアクソンの変性が起きてくることが知られ(二次性軸索変性)、アクソンの維持にも必要であることが知られている(6)。
臨床症状
生直後から遅くも1ヶ月程度までに眼振で気づかれることが多い。水平性眼振であることが多く、代償性の頭部振戦を認める場合がある。生後から半年程度までは筋緊張低下の症状を呈するが、原始反射の消失が遅れ、バビンスキー反射は半年を超えても陽性であり、やがて腱反射の亢進も明らかになり一次ニューロンの問題を示す。小脳症状としての企図振戦は1歳過ぎには、注意深く観察すると明らかであることが多い。また2歳頃にはアテトーゼ様の異常肢位が発現してくる(7)。このように中枢神経系の、運動、運動制御系、基底核のすべての症状が相次いで出現するのがこの疾患の特徴であるが、後年眼振は目立たなくなり、関節拘縮が進むと小脳症状も気づかれず、年長児で痙性と固縮をもつ脳性麻痺として診断されている例も多い。また乳幼児期の運動知的発達が正常で、学童期以降にゆっくりと退行する、X連鎖性痙性対麻痺の表現型をとることもある。通常10-20歳代を過ぎると症状の退行が始まり、平均寿命は30歳前後である。症状の退行と平行して、画像上の脳萎縮を認める。PLP1機能喪失変異の症例では、末梢神経障害を呈する。
検査・画像所見
検査所見では、聴性脳幹反応では、II波以降がまだらな中枢伝導時間を反映して消失する。末梢神経伝導速度は正常であるが、PLP1欠失など機能喪失型の変異による症例では、軽度低下する。血清生化学・尿所見に異常は認めない。髄液所見では、蛋白上昇を含め明らかな異常は認めない。脱髄疾患とは異なりmyelin basic proteinの値も正常範囲から正常上限であることが多い。MRI画像は極めて特徴的であり、T2強調画像では瀰漫性の白質の高信号およびT1強調画像で、皮質・白質のコントラストが消失(あるいは低下)していることが特徴的である。一般的に、T2強調像の信号では正常の新生児期よりも髄鞘化に乏しく年齢が長じても変化が乏しい(8)。T1強調画像上は、ゆっくりとした髄鞘化を認めるが、皮質・白質のコントラストが悪い。CT検査では、非特異的な大脳萎縮を認めるのみで診断的な情報は少ない。
遺伝子診断
PLP1遺伝子重複、点変異、遺伝子欠失など様々な変異が存在する。1PLP1遺伝子重複はほぼ半数(50-75%)の症例で認める。これは、定量的PCR法や間期核FISH法などにより正常の2倍量のPLP1の存在により確認できる。また、MLPA(Multiplex ligation-dependent probe amplification)やアレイCGH(microarray-based comparative genomic hybridization)などでも診断可能である。2割前後(15-25%)の患者では、PLP1タンパクコード領域やスプライズ部位の点変異による。アミノ酸置換型変異が最も頻度が高いが、他にスプライシング異常やナンセンス変異、挿入あるいは欠失なども認める(9)。変異の検出には、直接塩基配列決定法などを用いる。PLP1遺伝子全長の欠失は稀である(2%以下)。欠失は上記のいずれの方法でも検出される。PMD遺伝子診断は、変異の多様性を念頭に置き、異なる検査方法を組み合わせる必要がある。
2. 治療、ケア、
先天性白質形成不全症では、現在のところ根本的な治療がないため、各症状に対応した治療を行う。(以下はCQ4へ移行予定)
a. 精神運動発達遅滞
知的障害に、運動障害を伴うことからもっとも罹患率の多い、脳性麻痺児と同様の療育を受けることが実際的である。保健所あるいは診断を行った病院より、療育センターや病院のリハビリテーション科を紹介する。聴性脳幹反応が異常であるために、聴覚異常と判定される場合があるが、通常聴力に異常は認めない。言語理解まで獲得された場合、表出性言語能力よりも受容性言語理解がまさっている。脳性麻痺との違いは、発達退行が脳性麻痺よりもはっきりと現れる点にある。
b. てんかん
てんかんが、先天性白質形成不全の何割にみられるのか、系統的な報告はないが、恐らく10-20%の間と推定される。治療は発作のタイプにより、部分発作にはカルバマゼピン( 5-15 mg/kg, 2X)を第一選択とし、第二次選択薬としてはラモトリジン、トピラマート(4-10 mg/kg, 2X)、ゾニサミド(4-10 mg/kg, 2X)、バロプロ酸(15-40 mg/kg, 3X)、クロバザム(0.2-1 mg/kg, 2X)等のベンゾジアゼピン系抗痙攣薬を用いる。全般発作には第一選択薬バロプロ酸、フェノバルビタール (2-5 mg/kg, 1-2X)を用い、第二次選択薬としてはラモトリジン、トピラマート、ゾニサミド、クロバザム等のベンゾジアゼピン系抗痙攣薬を用いる。
c. ジストニア
全身性のジストニアに関してはエペリゾン(ミオナール)(1-4mg/kg, 3X), ジアゼパム(0.1-0.3mg/kg, 1-3X)、バクロフェン(リオレサール)(0.1-0.3-0.6mg/kg, 1-3X)、ダントロレンナトリウム(ダントリウム)(0.5mg/kg-3mg/kg, 2-3X)、ジサニジン(テルネリン)(0.05-0.1-0.15mg/kg, 1-3X)などフェノバルビタール(2-5 mg/kg, 1-2X)を用いる。局所性のジストニアでは、ボツリヌス毒素(1-3U/Kg)を用いる(最大3ヶ月毎)。
d. 股関節の痙性脱臼
大腿骨が内転・内旋・屈位になりやすいためにおこる。外転位保持夜間装具が必要となる場合がある。高度例では整形外科的な腸腰筋延長・切離術をおこなう。
e. 呼吸障害・摂食障害
喉頭咽頭機能不全のために、誤嚥性肺炎を起こしやすい。経口摂取が難しい症例では、経胃管あるいは胃瘻からの栄養補給が行われる。筋緊張亢進のために、胃食道逆流を伴う症例では、噴門形成術を併用する。
3. 食事・栄養
特に推奨される食事、栄養はない。栄養素のバランスにとれた食事が望ましいと考えられる。
4. 予後
自然歴の調査は存在しないが、10台から20代より退行がはじまると考えられている。
5. 鑑別診断
PMDL (PMD-like disease)はgap junction protein 12; GJA12 (connexin 47 ともよばれる) の異常による。臨床症状は、ほとんどの症例では、座位・歩行を獲得しておりPMDと比して最大の発達レベルは高いが、10歳前に退行が始まる例が多く、軸索への障害がより強いものと考えられている(17)。その他ミ先天性大脳白質形成不全症の疾患が鑑別診断となる。
6. 最近のトピック
- クルクミン;井上らは、クルクミン投与により、PMDモデルマウスの生存期間の延長を認め、治験が計画されている(10)。
- iPS; 下島、山本らはPLP1遺伝子重複を持つPMD患者からiPS細胞を樹立した(11)
また沼澤、岡野らはPLP1アミノ酸変異を持つ患者から、iPS細胞を樹立し、オリゴデンドロサイトに分化させ、小胞体ストレスの亢進や髄鞘化形成の低下を再現した(12)。 - 臍帯血移植;ウィシュニューらは2名のPMD患者に臍帯血移植を行い、若干の髄鞘化を認めたが、自然歴との差異は明らかではない (13)。
- 幹細胞移植;ヒト神経幹細胞を脳内に移植するフェイズ1の臨床研究が、米国で行われた。4例中3例で軽度の運動発達の改善をを認め、大きな有害事象は認めなかった(14)
(治療のCQでもう少し詳しく記述予定)
図の説明
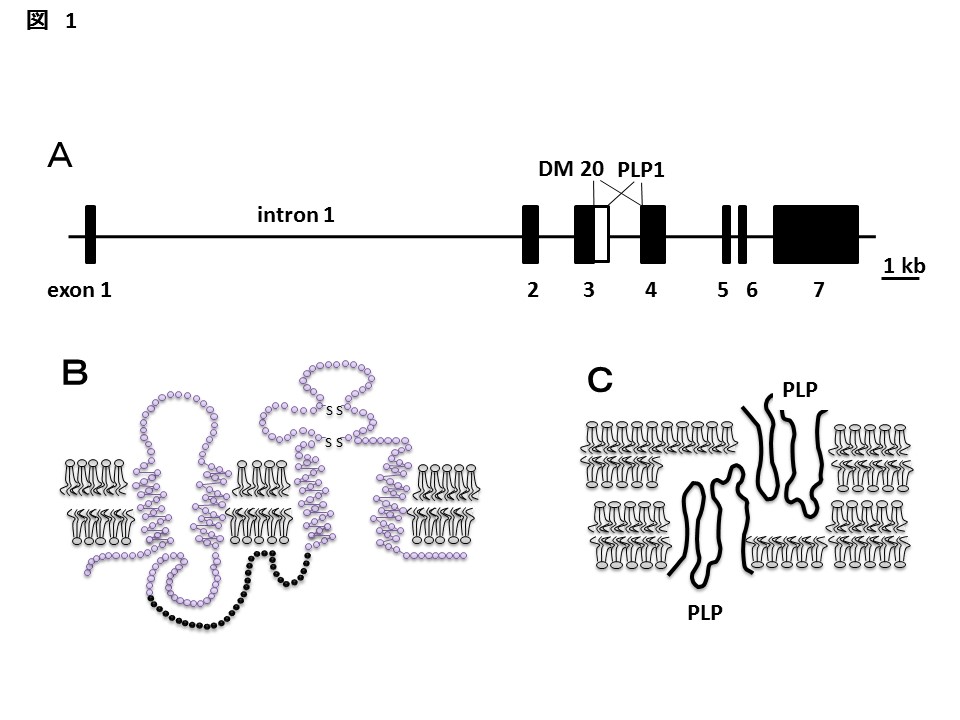
- PLP(proteolipid proein)1遺伝子の構造。7つのエクソンからなり、イントロン1は巨大であり、オリゴデンドロサイト特異的な発現に関わる。DM20ではエクソン3/イントロン3の境界部分はPLP1に比べて105bp上流部分となる。
- 予想されるPLP1の構造。4回膜貫通型膜蛋白質であり、DM 20は細胞内の黒丸の部分を欠く。
- ミエリン上でのPLP1の予想される配置。ミエリンの接着・緻密化に関わると考えられている。
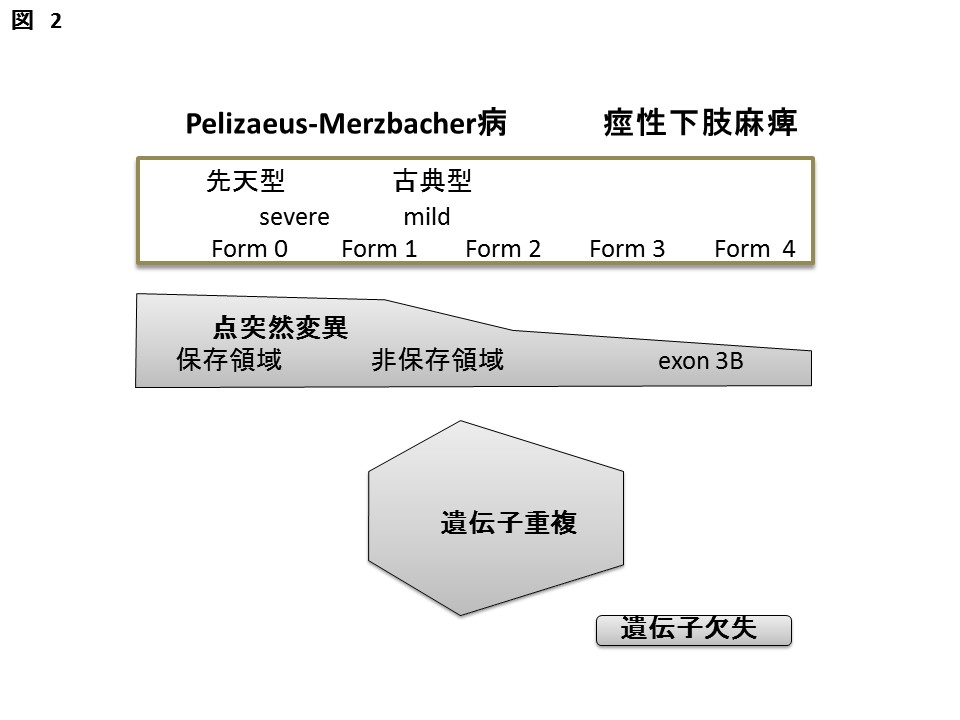
PLP1遺伝子異常と重症度の対応。(FORM 0; 定頸が見られない、FORM 1; 定頸は獲得するが、座位はとれない。FORM 2; 座位は獲得するが、支持歩行を獲得しない、FORM 3; 支持歩行は獲得するが自立歩行に至らない、FORM 4;自立歩行を獲得する。)従来の分類との対比ではFORM Oは先天型に、FORM 1および2は古典型に、FORM 3は軽症型PMDに、FORM 4はX連鎖性痙性下肢麻痺に対応している。遺伝子変異との対比では、アミノ酸置換は、保存された残基ではFORM 0、非保存残基ではFORM 1-2、エクソン3後半の変異では、FORM 3-4となることが多い。重複ではFORM 1-2、欠失型変異(欠失およびナンセンス変異による短い蛋白生成)ではFORM 3-4をとる症例が多い
引用文献(末尾に記載のないものは全てエビデンスレベル6)
- Seitelberger F. Neuropathology and genetics of Pelizaeus-Merzbacher disease. Brain pathology. 1995;5(3):267-73.
- Hudson LD, Puckett C, Berndt J, Chan J, Gencic S. Mutation of the proteolipid protein gene PLP in a human X chromosome-linked myelin disorder. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(20):8128-31.
- Trofatter JA, Dlouhy SR, DeMyer W, Conneally PM, Hodes ME. Pelizaeus-Merzbacher disease: tight linkage to proteolipid protein gene exon variant. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(23):9427-30.
- Inoue K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics. 2005;6(1):1-16.
- Numata Y, Gotoh L, Iwaki A, Kurosawa K, Takanashi J, Deguchi K, et al. Epidemiological, clinical, and genetic landscapes of hypomyelinating leukodystrophies. J Neurol. 2014;261(4):752-8.
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280(5369):1610-3.
- Osaka H, Inoue K. Pathophysiology and emerging therapeutic strategies in Pelizaeus–Merzbacher disease. Expert Opinion on Orphan Drugs. 2015;3(12):1447-59.
- Takanashi J, Sugita K, Tanabe Y, Nagasawa K, Inoue K, Osaka H, et al. MR-revealed myelination in the cerebral corticospinal tract as a marker for Pelizaeus-Merzbacher's disease with proteolipid protein gene duplication. AJNR American journal of neuroradiology. 1999;20(10):1822-8.
- Hobson GM, Kamholz J. PLP1-Related Disorders. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, et al., editors. GeneReviews(R). Seattle (WA)1993.
- Morimura T, Numata Y, Nakamura S, Hirano E, Gotoh L, Goto YI, et al. Attenuation of endoplasmic reticulum stress in Pelizaeus-Merzbacher disease by an anti-malaria drug, chloroquine. Experimental biology and medicine. 2014;239(4):489-501.
- Shimojima K, Inoue T, Imai Y, Arai Y, Komoike Y, Sugawara M, et al. Reduced PLP1 expression in induced pluripotent stem cells derived from a Pelizaeus-Merzbacher disease patient with a partial PLP1 duplication. Journal of human genetics. 2012;57(9):580-6.
- Numasawa-Kuroiwa Y, Okada Y, Shibata S, Kishi N, Akamatsu W, Shoji M, et al. Involvement of ER stress in dysmyelination of Pelizaeus-Merzbacher Disease with PLP1 missense mutations shown by iPSC-derived oligodendrocytes. Stem cell reports. 2014;2(5):648-61.
- Wishnew J, Page K, Wood S, Galvin L, Provenzale J, Escolar M, et al. Umbilical cord blood transplantation to treat Pelizaeus-Merzbacher Disease in 2 young boys. Pediatrics. 2014;134(5):e1451-7. エビデンスレベル5
- Gupta N, Henry RG, Strober J, Kang SM, Lim DA, Bucci M, et al. Neural stem cell engraftment and myelination in the human brain. Science translational medicine. 2012;4(155):155ra37. エビデンスレベル5
文献検索
PubMed
- "pelizaeus-merzbacher disease"[MeSH Terms] OR ("pelizaeus-merzbacher"[All Fields] AND "disease"[All Fields]) OR "pelizaeus-merzbacher disease"[All Fields] OR ("pelizaeus"[All Fields] AND "merzbacher"[All Fields] AND "disease"[All Fields]) OR "pelizaeus merzbacher disease"[All Fields]
581件 - ("proteolipids"[MeSH Terms] OR "proteolipids"[All Fields] OR "proteolipid"[All Fields]) AND ("proteins"[MeSH Terms] OR "proteins"[All Fields] OR "protein"[All Fields])
7430件
医中誌
- (Pelizaeus-Merzbacher病/TH or pelizaeus-merzbacher/AL) 317 件
- (Proteolipids/TH or proteolipid/AL) and (タンパク質/TH or protein/AL) 530 件









