








Guidelines on Hereditary Leukodystrophies
Pelizaeus-Merzbacher disease (PMD, HLD1, OMIM#312080)
Disease description: Pelizaeus-Merzbacher disease is a form of hypomyelination of the central nervous system caused by an abnormality in the gene that encodes proteolipid protein 1. Because it is an X-linked autosomal recessive inherited disease, with very few exceptions it occurs only in boys. It causes congenital nystagmus and hypotonia in infancy, after which formation, rigidity, and cerebellar ataxia start to appear. In severe cases, babies never develop the ability to hold up their heads, but children with mild symptoms may learn to walk. Intellectual development is better than motor development.
Treatment: Umbilical cord blood transplantation and stem cell transplantation have been attempted in a few patients but without any dramatic improvement. At this point, symptomatic therapy is provided.
1. Overview
Definition
Pelizaeus-Merzbacher disease (PMD) was first described by German doctor Friedrich Pelizaeus in 1885 and German pathologist Ludwig Merzbacher in 1910 and subsequently established as a pathological and clinical syndrome by Seitelberger in 1970 (1). It is a form of hypomyelination of the central nervous system that occurs in boys that is pathologically characterized by the disappearance or extreme reduction of the myelin sheath in the white matter with neuronal conservation, with no obvious demyelinating changes, and patches of conserved myelin with a tigroid appearance. A subsequent genetic analysis revealed that a mutation in the gene encoding proteolipid protein 1 (PLP1) is mainly responsible for the disease (2, 3). PLP1, the main constituent molecule of the myelin protein, is expressed in oligodendrocytes of the central nervous system; therefore, the disease is a form of dysmyelination with symptoms limited to the central nervous system (4).
Epidemiology
The incidence of PMD in Japan is 0.26 per 100,000 births, similar to the levels reported in the United States and Germany (5).
Etiology and pathophysiology
Myelin, which provides electrical insulation around the axons of the central nervous system, consists of approximately 80% fat and 20% protein. Around 50% of this protein consists of proteolipid protein 1 (PLP1), the most important constituent protein in quantitative terms. The PLP1 gene consists of seven exons encoding two proteins, PLP1 and DM20, both of which are believed to have four transmembrane domains (Figures 1A, 1B) (3). DM20 lacks part of PLP1 (35 amino acids corresponding to the second half of exon 3). During fetal development, its expression in a range of organs is dominant over that of PLP1; in lower animals, only DM20 orthologues are present. PLP1 expression is upregulated after birth but limited to central oligodendrocytes. In PLP1 knockout mice, the myelin sheath loses its compact layered structure, space opens up between the myelin layers, and central conduction time is prolonged, suggesting that PLP1 may contribute to stabilizing myelin membrane binding (Figure 1C) (5). Axon degeneration (secondary axonopathy) is known to occur in mice as well as patients who have lost this gene, showing that it is required for axon maintenance (6).
Clinical symptoms
Nystagmus is usually noticed by around 1 month after birth. In most cases, it occurs horizontally and compensatory head bobbing may occur. Symptoms of dystonia usually appear by around 6 months after birth, but the loss of primitive reflexes is delayed, with the Babinski reflex still present after >6 months; after a while, enhanced tendon reflexes start to appear, indicating a primary neuron problem. The cerebellar symptom of intention tremor is often noticeable on close observation after 1 year of age. Athetoid abnormal limb positions start to appear at around 2 years of age (7). A characteristic of this disease is that all central nervous system motor, motor control, and basal ganglia symptoms appear one after the other. Over time, the nystagmus becomes less obvious, and as joint contracture progresses, cerebellar symptoms become less noticeable, leading to spasticity and rigidity at age 5 or 6 years frequently being misdiagnosed as cerebral palsy. Psychomotor development is normal during infancy but gradually regresses after around age 6 years, and the disease may also exhibit the phenotype of X-linked spastic paraplegia. Symptom regression usually starts during the teens or twenties, while the mean lifespan is around 30 years of age. Imaging signs of brain atrophy develop in parallel with symptom regression. Patients with a PLP1 loss-of-function mutation also exhibit peripheral neuropathy.
Imaging and other investigations
Tests show that waves II–V of the auditory brainstem response are lost, reflecting patchy central conduction time. Peripheral nerve conduction velocity is usually normal, but it decreases slightly in patients with loss-of-function mutations such as PLP1 deletion. Serum biochemistry and urinalysis results are unremarkable. Cerebrospinal fluid test results, including protein content, do not reveal any obvious abnormalities. Unlike demyelinating disorders, in most cases the level of myelin basic protein is within or above normal limits. MRI findings are highly characteristic, typically displaying diffuse hyperintensity of the white matter on T2-weighted imaging and loss (or reduction) of contrast between the cortex and white matter on T1-weighted imaging. On T2-weighted imaging, the signal T2 indicates poor myelination compared with healthy neonates with little change over time (8). On T1-weighted imaging, myelination occurs slowly, but the contrast between the cortex and the white matter is poor. The only characteristic sign on computed tomography is cerebral atrophy, and it provides little diagnostic information.
Genetic diagnosis
A range of different mutations may occur in the PLP1 gene, including duplication, point mutation, and gene deletion. A 1PLP1 duplication is present in around half (50–75%) of patients. This can be confirmed by quantitative polymerase chain reaction (PCR) or interphase nuclear fluorescence in situ hybridization, which demonstrate the presence of twice the normal amount of PLP1. It can also be diagnosed using techniques such as multiplex ligation–dependent probe amplification and microarray-based comparative genomic hybridization. In around 15–25% of patients, the disease is caused by a point mutation in the PLP1 protein–coding region or splicing site. Amino acid substitution mutations are the most common form, but other splicing defects and nonsense mutations, insertions, or deletions are also seen (9). Direct sequencing or a similar method is used to detect the mutation. Deletion of the entire PLP1 gene is rare (£2%). Deletions can also be detected by any of the methods described above. Given the diversity of possible mutations, the genetic diagnosis of PMD must involve a combination of several different screening methods.
2. Treatment and care
Since there is currently no cure for congenital cerebralhypomyelination, symptomatic therapy is provided. (The information below is to be moved to CQ4)
a. Delayed psychomotor development
Because a motor disability is present alongside intellectual disability, in practice many children receive the same special needs care and education as that provided for children with cerebral palsy, the most common similar condition. They are referred by a public health center or the hospital where they are diagnosed to an institution for children with special needs or a hospital rehabilitation department. Because their auditory brainstem response is abnormal, they may be diagnosed as hearing-impaired when in fact their hearing is normal. If they become able to understand speech, their passive language comprehension exceeds their expressive language ability. Unlike cerebral palsy, developmental regression is clearly evident.
b. Epilepsy
Although there has been no statistical study of the proportion of patients with congenital cerebral hypomyelination who suffer from epilepsy, it is estimated as possibly occurring in 10–20% of cases. The treatment depends on the type of seizure involved. For partial seizures, carbamazepine (5–15 mg/kg twice daily) is the first-line drug, while second-line drugs include lamotrigine, topiramate (4–10 mg/kg twice daily), zonisamide (4–10 mg/kg twice daily), valproic acid (15–40 mg/kg 3 times daily), or a benzodiazepine anticonvulsant such as clobazam (0.2–1 mg/kg twice daily). For full seizures, the first-line drug is valproic acid or phenobarbital (2–5 mg/kg 1–2 times daily), and the drugs used as second-line treatments include lamotrigine, topiramate, zonisamide, and benzodiazepine anticonvulsants such as clobazam.
c. Dystonia
Generalized dystonia is treated with eperisone (1–4 mg/kg 3 times daily), diazepam (0.1–0.3 mg/kg 1–3 times daily), baclofen (0.1–0.3–0.6 mg/kg 1–3 times daily), dantrolene (0.5–3 mg/kg 2–3 times daily), tizanidine (0.05–0.1–0.15 mg/kg 1–3 times daily), or phenobarbital (2–5 mg/kg 1–2 times daily). Botox (1–3 U/kg) is used to treat localized dystonia (no more than once every 3 months).
d. Spastic dislocation of the hip
This occurs because the femur is readily adducted, internally rotated, and flexed. It may be necessary to use a night-time brace to retain it in the abducted position. In severe cases, orthopedic surgery may be performed to extend or transect the iliopsoas muscle.
e. Respiratory and eating disorders
Laryngopharyngeal dysfunction may make patients prone to aspiration pneumonia. Patients who have difficulty ingesting food by mouth may be fed via a nasogastric tube or a gastrostoma; if gastroesophageal reflux is present due to muscle hypertonia, then fundoplication is also performed.
3. Diet and nutrition
No particular diet or type of nutrition is recommended. A nutritionally balanced diet is desirable.
4. Prognosis
There has been no survey of the natural history of PMD, but regression is believed to start during the teens or twenties.
5. Differential diagnosis
PMD-like disease is caused by a defect in gap junction protein 12 (GJA12, also known as connexin 47). In terms of clinical symptoms, most patients become able to sit up and walk, exceeding the maximum level of development seen in PMD, but regression usually starts before age 10 and axonal damage is believed to be worse (17). Other non-congenital diseases causing cerebral leukodystrophy must also be included in the differential diagnosis.
6. Recent topics
- Curcumin: Inoue et al. observed that feeding curcumin to PMD model mice prolonged their survival; a clinical trial is planned (10).
- iPS: Shimojima, Yamamoto et al. established an iPS cell line from PMD patients with PLP1 duplication (11).
Numasawa, Okuno et al. also established an iPS cell line from patients with a PLP1 amino acid mutation, induced them to differentiate into oligodendrocytes, and reproduced increased endoplasmic reticulum stress and reduced myelin formation (12). - Umbilical cord blood transplantation: Wishnew et al. transplanted umbilical cord blood into 2 PMD patients and observed some myelination, but there was no obvious difference from that of the natural history of the disease (13).
- Stem cell transplantation: A Phase 1 clinical study of the transplantation of human neural stem cells into the brain has been carried out in the United States. Slightly improved motor development was evident in three-quarters of patients, and no major adverse events occurred (14).
(This is to be described in slightly more detail in the Treatment CQ.)
Figure legends
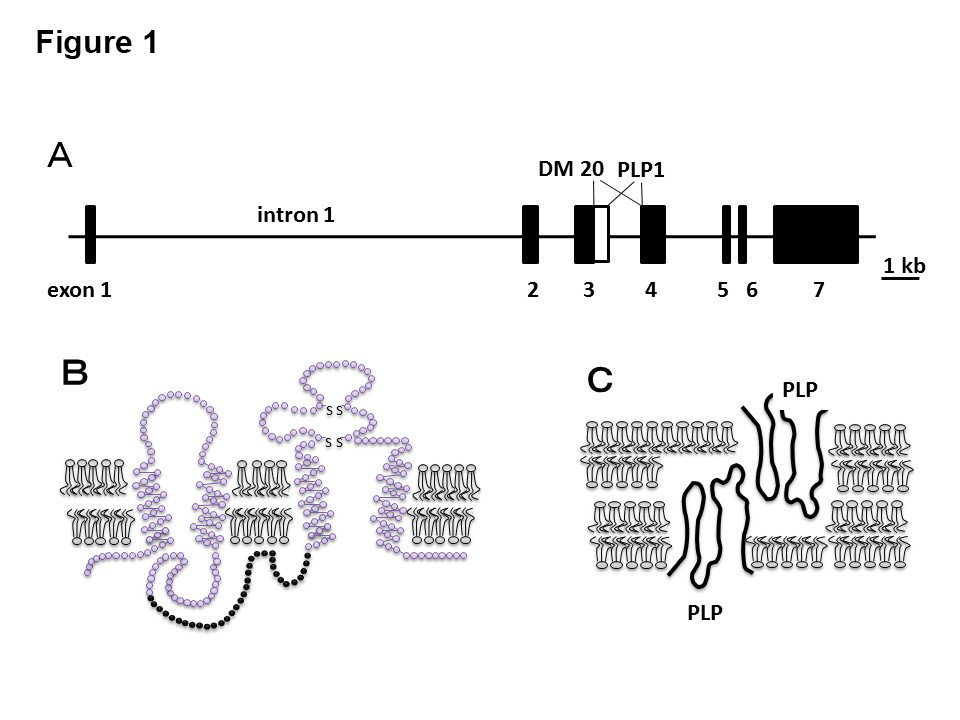
- The proteolipid protein 1 (PLP1) gene consists of 7 exons; intron 1 is massive and involved in oligodendrocyte-specific expression. In DM20, the boundary area between exon 3 and intron 3 is 105 bp upstream compared with PLP1.
- The expected structure of PLP1, a membrane protein with 4 transmembrane domains; in DM20, the intracellular section denoted by black circles is absent.
- The expected position of PLP1 in myelin. It is believed to be involved in myelin adhesion and densification.
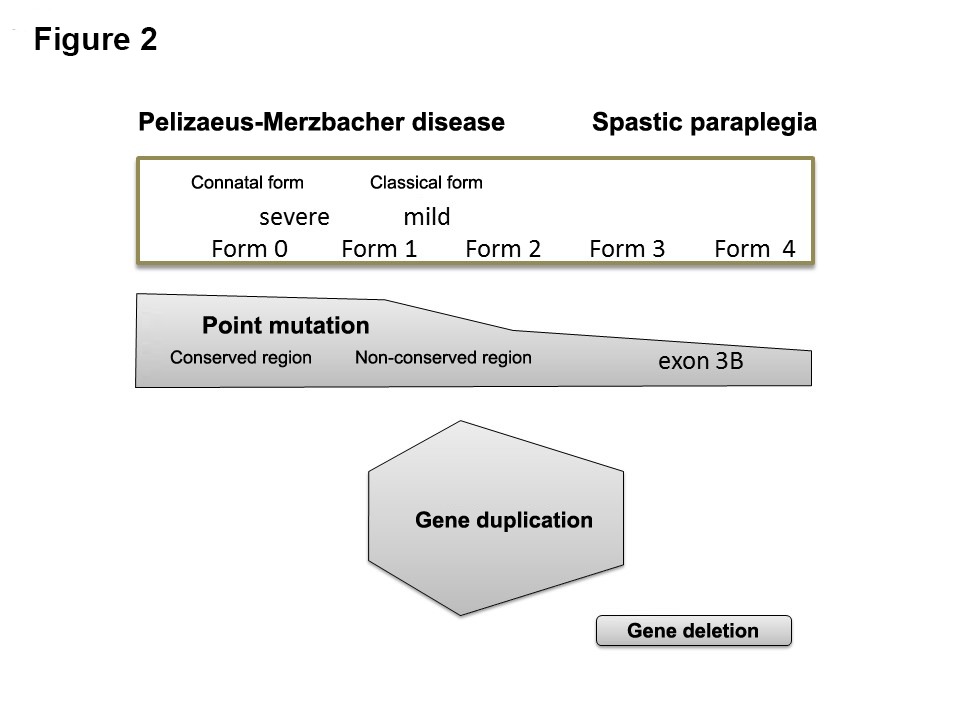
Correspondence between PLP1 mutations and severity. FORM 0: unable to lift head. FORM 1: Able to lift head but not sit up. FORM 2: Able to sit up but not walk even with support. FORM 3: Able to walk with support but not unaided. FORM 4: Able to walk unaided. In comparison with the conventional classification, FORM 0 corresponds to the connatal form, FORM 1–2 to the classical form, FORM 3 to mild PMD, and FORM 4 to X-linked spastic paraplegia. In terms of correspondence with gene mutations, amino acid substitutions are usually FORM 0 for conserved residues and FORM 1–2 for non-conserved residues, while mutations in the distal part of exon 3 usually cause FORM 3–4. Most patients with duplications exhibit FORM 1–2, whereas most of those with deletion-type mutations (short protein synthesis due to a deletion or nonsense mutation) exhibit FORM 3–4.
References
(Unless otherwise noted at the end, all are evidence level 6.)
- Seitelberger F. Neuropathology and genetics of Pelizaeus-Merzbacher disease. Brain Pathol 1995; 5(3): 267-273.
- Hudson LD, Puckett C, Berndt J, Chan J, Gencic S. Mutation of the proteolipid protein gene PLP in a human X chromosome-linked myelin disorder. Proc Natl Acad Sci U S A 1989; 86(20): 8128-8131.
- Trofatter JA, Dlouhy SR, DeMyer W, Conneally PM, Hodes ME. Pelizaeus-Merzbacher disease: tight linkage to proteolipid protein gene exon variant. Proc Natl Acad Sci U S A 1989; 86(23): 9427-9430.
- Inoue K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics 2005; 6(1): 1-16.
- Numata Y, Gotoh L, Iwaki A, Kurosawa K, Takanashi J, Deguchi K, et al. Epidemiological, clinical, and genetic landscapes of hypomyelinating leukodystrophies. J Neurol 2014; 261(4): 752-758.
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 1998; 280(5369): 1610-1613.
- Osaka H, Inoue K. Pathophysiology and emerging therapeutic strategies in Pelizaeus–Merzbacher disease. Exp Opin Orphan Drugs 2015; 3(12): 1447-1459.
- Takanashi J, Sugita K, Tanabe Y, Nagasawa K, Inoue K, Osaka H, et al. MR-revealed myelination in the cerebral corticospinal tract as a marker for Pelizaeus-Merzbacher's disease with proteolipid protein gene duplication. Am J Neuroradiol 1999; 20(10): 1822-1828.
- Hobson GM, Kamholz J. PLP1-related disorders. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, et al., editors. GeneReviews(R). Seattle WA: University of Washington, Seattle; 1993.
- Morimura T, Numata Y, Nakamura S, Hirano E, Gotoh L, Goto YI, et al. Attenuation of endoplasmic reticulum stress in Pelizaeus-Merzbacher disease by an anti-malaria drug, chloroquine. Exp Biol Med 2014; 239(4): 489-501.
- Shimojima K, Inoue T, Imai Y, Arai Y, Komoike Y, Sugawara M, et al. Reduced PLP1 expression in induced pluripotent stem cells derived from a Pelizaeus-Merzbacher disease patient with a partial PLP1 duplication. J Hum Genet 2012; 57(9): 580-586.
- Numasawa-Kuroiwa Y, Okada Y, Shibata S, Kishi N, Akamatsu W, Shoji M, et al. Involvement of ER stress in dysmyelination of Pelizaeus-Merzbacher Disease with PLP1 missense mutations shown by iPSC-derived oligodendrocytes. Stem Cell Repts 2014; 2(5): 648-661.
- Wishnew J, Page K, Wood S, Galvin L, Provenzale J, Escolar M, et al. Umbilical cord blood transplantation to treat Pelizaeus-Merzbacher Disease in 2 young boys. Pediatrics 2014; 134(5): e1451-7.(5)
- Gupta N, Henry RG, Strober J, Kang SM, Lim DA, Bucci M, et al. Neural stem cell engraftment and myelination in the human brain. Sci Transl Med 2012; 4(155): 155ra37.(5)
Literature search
PubMed
- "pelizaeus-merzbacher disease"[MeSH Terms] OR ("pelizaeus-merzbacher"[All Fields] AND "disease"[All Fields]) OR "pelizaeus-merzbacher disease"[All Fields] OR ("pelizaeus"[All Fields] AND "merzbacher"[All Fields] AND "disease"[All Fields]) OR "pelizaeus merzbacher disease"[All Fields]
581 results - ("proteolipids"[MeSH Terms] OR "proteolipids"[All Fields] OR "proteolipid"[All Fields]) AND ("proteins"[MeSH Terms] OR "proteins"[All Fields] OR "protein"[All Fields])
7430 results
Igaku Chūō Zasshi
- (Pelizaeus-Merzbacher病/TH or pelizaeus-merzbacher/AL) 317 results
- (Proteolipids/TH or proteolipid/AL) and (タンパク質/TH or protein/AL) 530 results
Table 1. Classification of congenital cerebral leukodystrophies
Main diseases exhibiting congenital hypomyelination of the central nerves
| Disease | Inheritance pattern | Locus | Causative gene |
|---|---|---|---|
| Pelizaeus-Merzbacher disease | X-linked recessive | Xq22 | PLP1 |
| Pelizaeus-Merzbacher–like disease 1 | Autosomal recessive | 1q42.13 | GJC2 |
| Hypomyelination with atrophy of the basal ganglia and cerebellum | Autosomal dominant | 19p13.3 | TUBB4A |
| Chromosome 18q deletion syndrome | Chromosomal | 18q22-qter | MBP |
| Allan-Herndon-Dudley syndrome | X-linked recessive | Xq13.2 | SLC16A2 |
| Hsp60 chaperonopathy | Autosomal recessive | 2q33.1 | HSPD1 |
| Salla disease | Autosomal recessive | 6q13 | SLC17A5 |
| Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum | Autosomal recessive | 12q23.3 | POLR3B |
Diseases exhibiting congenital hypomyelination of the central and peripheral nerves
| Disease | Inheritance pattern | Locus | Causative gene |
|---|---|---|---|
| Hypomyelination with congenital cataract | Autosomal recessive | 7p15.3 | FAM126A |
| Hypomyelination with ataxia and hypodontia | Autosomal recessive | 10q22.3 | POLR3A |
| Peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease | Autosomal dominant | 22q13 | SOX10 |
PLP1, proteolipid protein 1; GJC2, gap junction protein; TUBB 4A, tubulin β-4A; MBP, myelin basic protein; POLR3A, B, two subunits (A and B) of RNA polymerase III; SLC16A2, solute carrier family 16 (monocarboxylic acid transporter), member 2 encoding triiodothyronine, T3, and thyroxine, T4 transporter, monocarboxylate transporter 8 (MCT8); HSPD1, encodes heat shock 60-kD protein, FAM126A, family with sequence similarity 126, member A, DRCTNNB1A; SOX 10, sry-related hmg-box gene 10