




遺伝性白質疾患ガイドライン
CQ2 大脳白質形成不全症はどのように画像診断しますか
大脳白質形成不全症の診断にMRI検査を行うことが強く勧められる。(推奨グレードA)
白質全体の淡いT2高信号が画像上の特徴である。疾患によっては特徴的な追加画像所見を呈し、診断の根拠となりうる。
解説
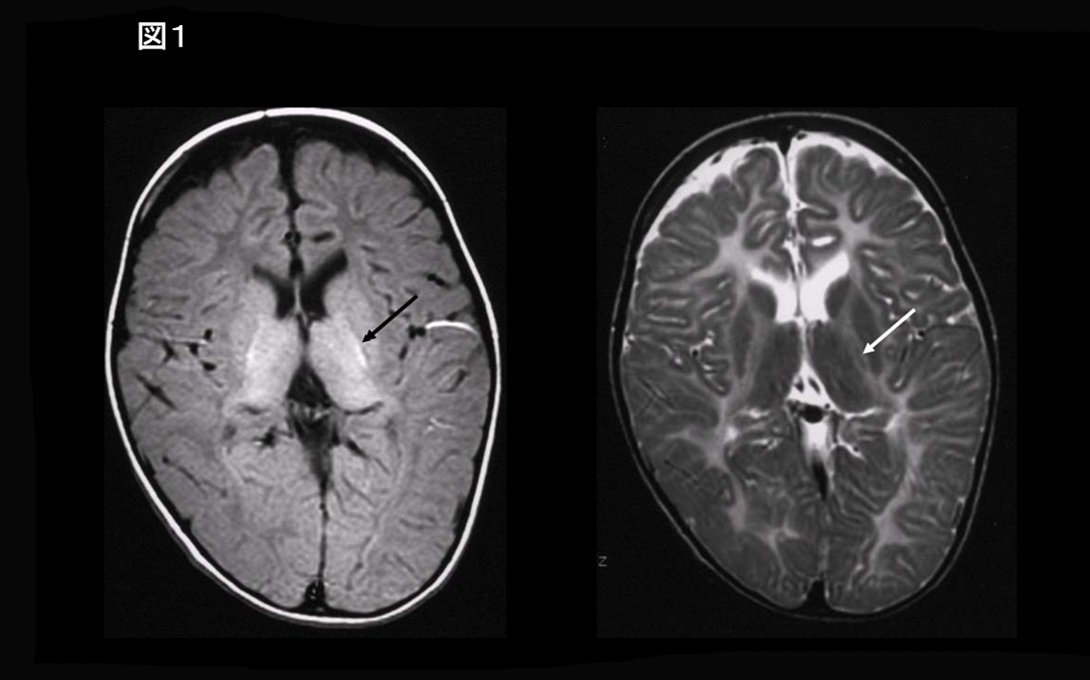
大脳白質形成不全症は髄鞘の形成障害ないし遅延を呈する一群であり、髄鞘化の未熟な新生児期の画像(白質は皮質に比べてT1低信号、T2高信号)と近似する。T2強調画像では白質全体が淡い高信号を呈するか、早期に髄鞘化を生じる部位(内包後脚、視放線、皮質脊髄路など)にのみ髄鞘化を反映した低信号を示す(図1)1-3)。髄鞘化に伴うMR信号変化はT2低信号に先立ちT1高信号を呈すことが知られている。髄鞘形成の程度によりT1強調画像は白質全体の淡い低信号、早期に髄鞘化の生じる部位のみの淡い高信号、広範な淡い高信号まで種々の信号を呈しうる。MRI 所見から大脳白質形成不全症と確定するには、白質信号異常が、6か月以上の間隔で不変であること、少なくとも1回は1歳時以降の MRI で評価することが必要とされる。2歳以降で MRI 上重度の画像所見が認められる場合は、大脳白質形成不全症の可能性が高い2, 3)。大脳白質形成不全症に分類される疾患の画像診断のフローチャートについて図2に記載し概説する。
次いで、各疾患の画像所見につき説明を加える。
大脳白質形成不全症の画像診断フローチャート(図2)
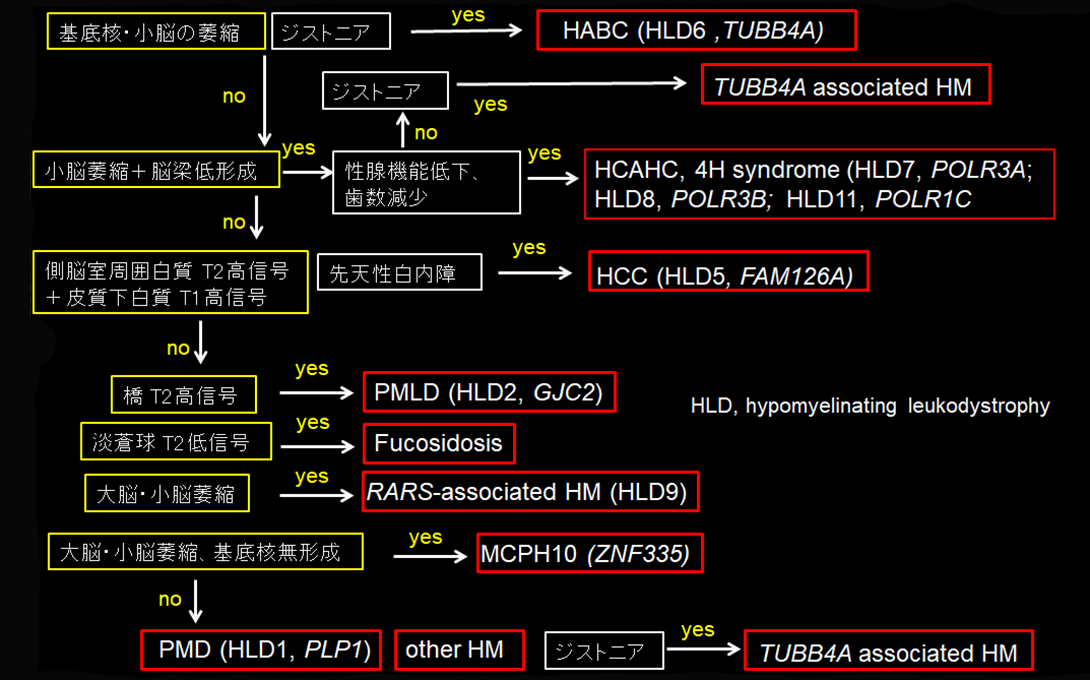
T2強調画像で白質全体が淡い高信号を呈し、大脳白質形成不全症が疑われた場合の診断フローチャートを概説する。図の黄色枠は画像所見、白枠は臨床症状、赤枠が診断である。
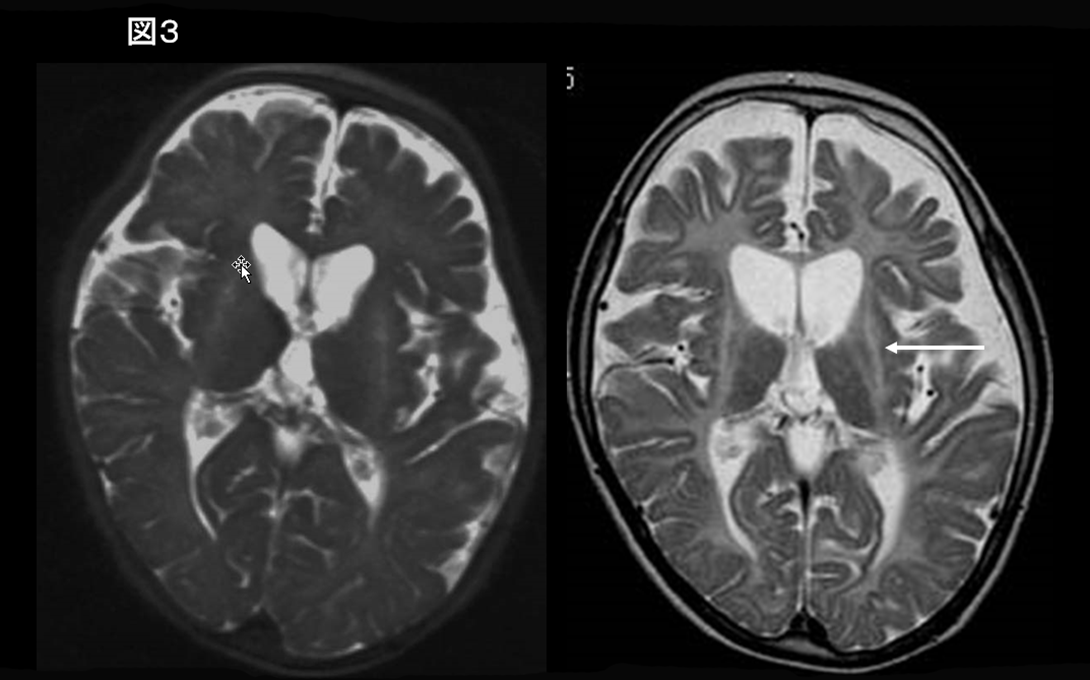
MRI画像上、大脳白質形成不全に加えて基底核・小脳の萎縮が認められる場合(図3)、Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC, HLD6) がまず疑われる。臨床症状としてジストニアに代表される錐体外路症状を呈することが多く、原因遺伝子TUBB4Aの検索を進める。
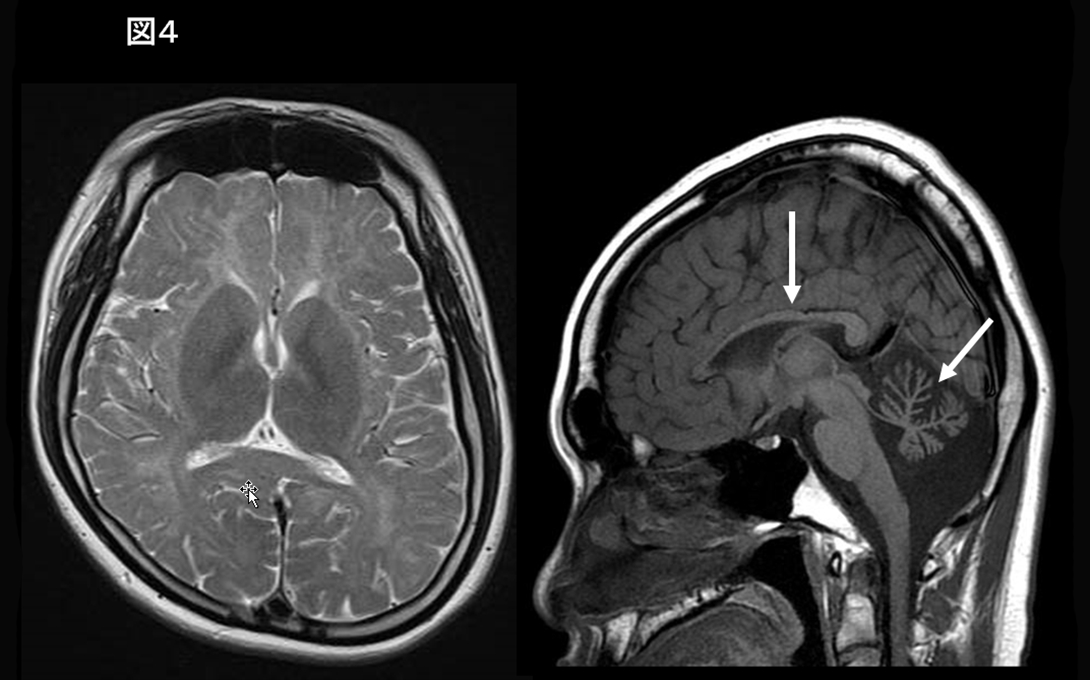
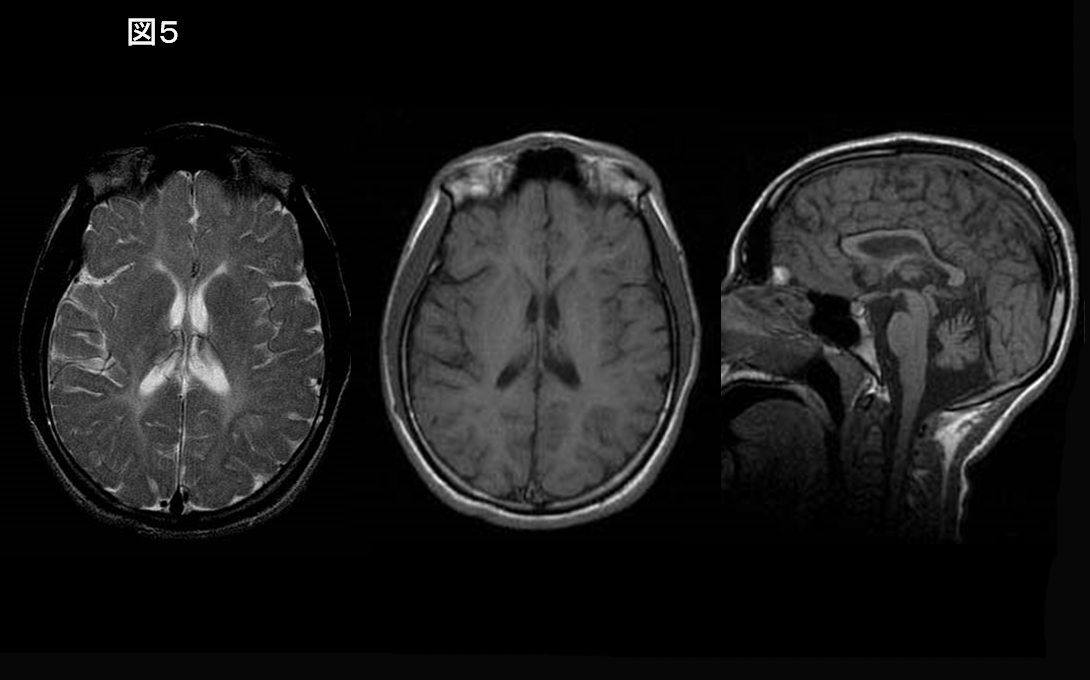
MRI画像上、基底核は正常であり、小脳萎縮・脳梁のひ薄化を認める(hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum [HCAHC])場合(図4)、Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome)が疑われる。性腺機能・歯数のチェックが必須であり、原因遺伝子POLR3A, 3B, 1Cの検索を進める。HCAHCの所見を呈し基底核に異常がない場合も、臨床的にジストニアを認める場合(図5)は TUBB4A-associated hypomyelinating disorder が疑われる。
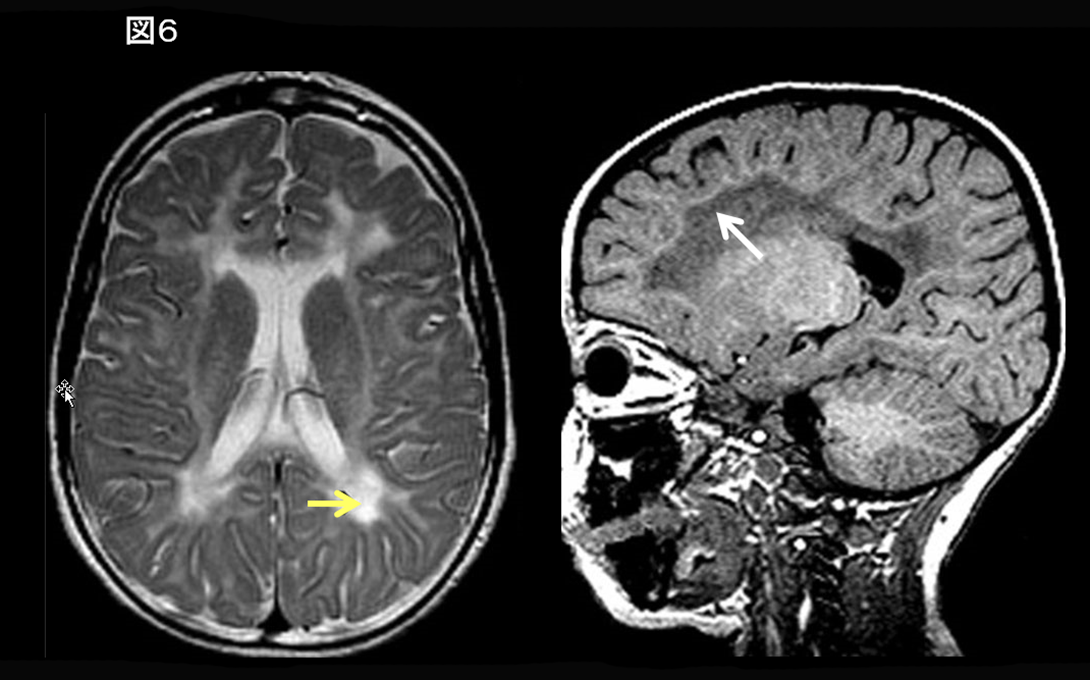
基底核・小脳所見を欠き、側脳室周囲に著明なT2高信号(大脳白質形成不全のT2高信号よりさらに異常高信号)、皮質下白質にT1高信号を認める場合(図6)、Hypomyelination and congenital cataract (HCC, HLD5) が疑われる。眼科検索(白内障)は必須であり、FAM126A遺伝子の検索を進める。
上記の当てはまらない場合、以下の画像所見に注目する。
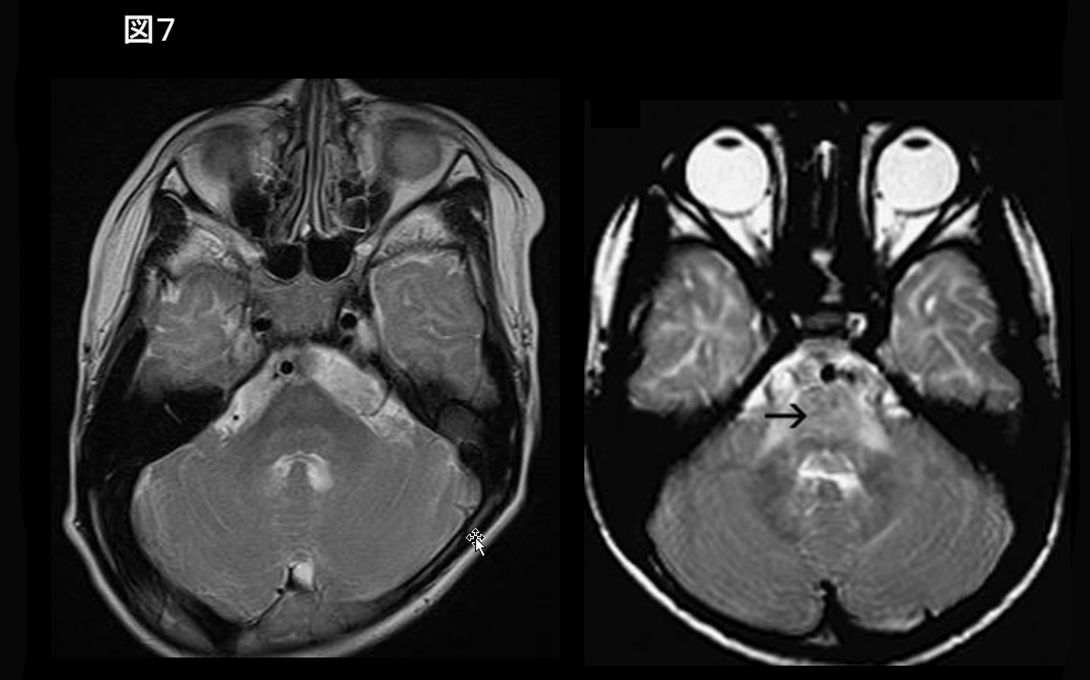
橋の著明なT2高信号を認める場合(図7)、Pelizaeus-Merzbacher-like disease (PMLD, HDL2, GJC2遺伝子異常) が疑われる。
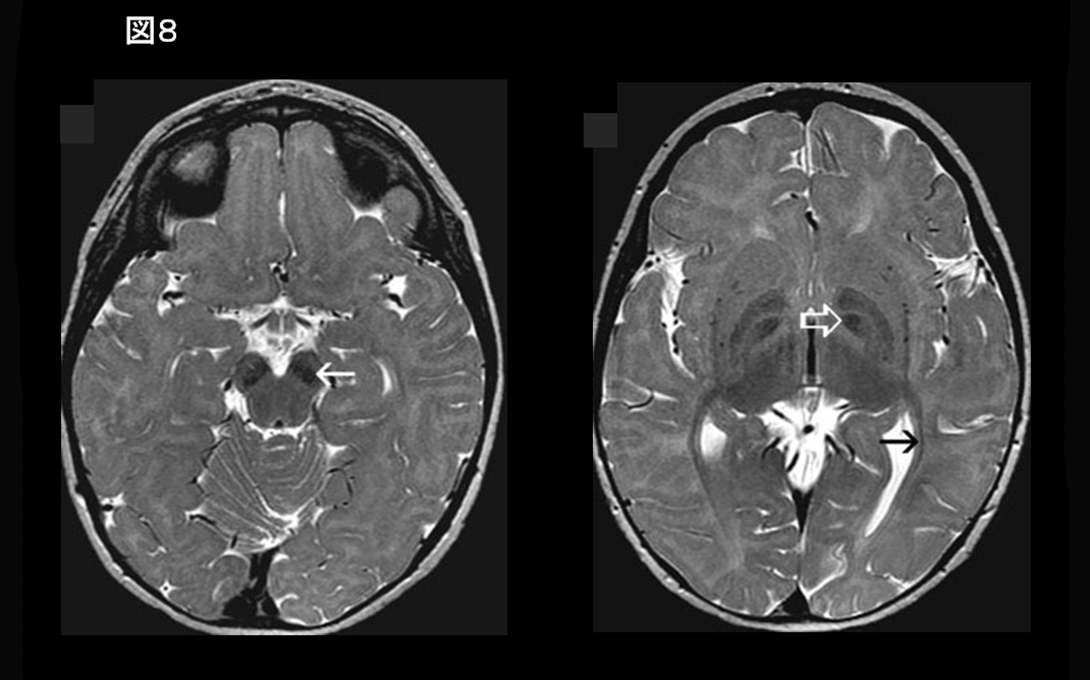
淡蒼球の著名なT2低信号を認める場合(図8)、Fucosidosisが疑われる。
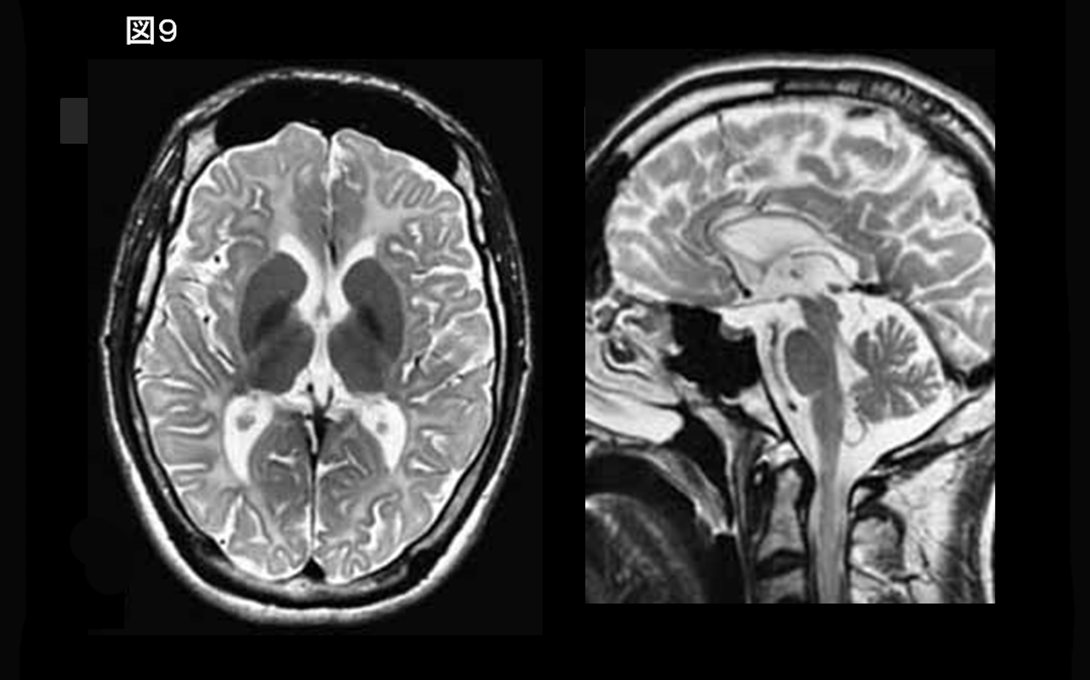
大脳・小脳の萎縮を認める場合(図9)、Hypomyelinating leukodystrophy-9 (HLD9, RARS遺伝子異常)が疑われる。
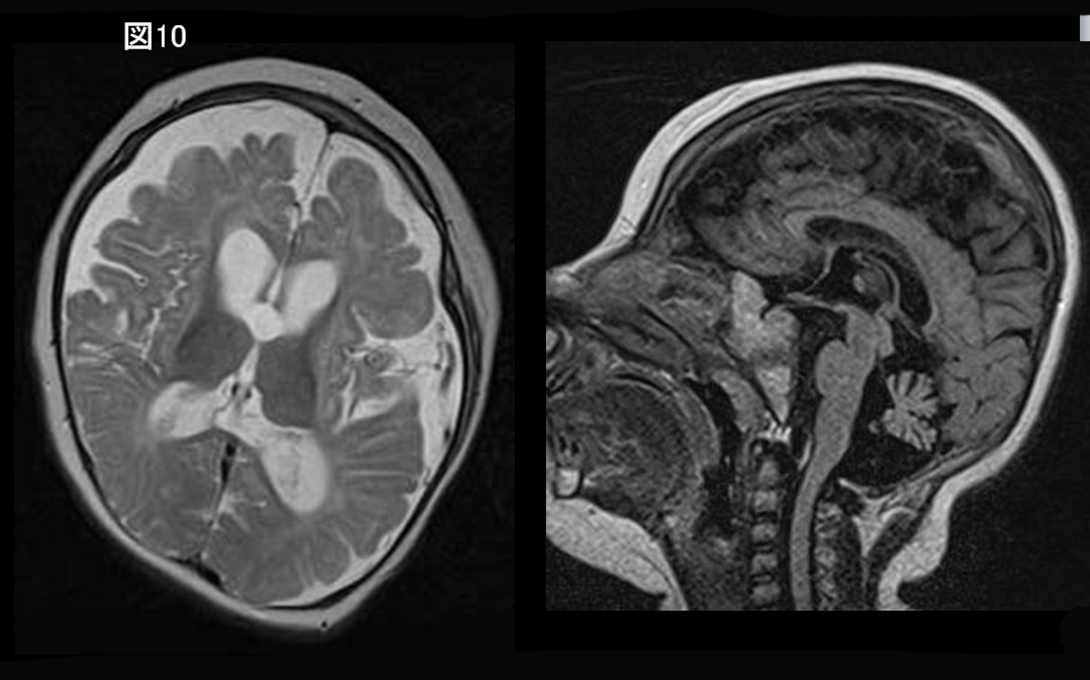
大脳・小脳の萎縮、脳幹低形成、基底核の無形性を認める場合(図10)、MCPH10 (ZNF335遺伝子異常) が疑われる。
上記に合致しない場合、Pelizaeus-Merzbacher disease (PMD, HLD1) を含め、他の大脳白質形成不全症の診断を進める。画像上、大脳白質形成不全の所見のみであっても、臨床的にジストニアを認める場合は TUBB4A-associated hypomyelinating disorder が疑われる。
各論
1). Pelizaeus-Merzbacher disease (PMD, HLD1, OMIM#312080)
Pelizaeus-Merzbacher diseaseは髄鞘の主たる構成蛋白であるproteolipid protein (PLP1) 遺伝子異常による伴性劣性遺伝疾患であり、先天性眼振、頭部振戦、痙性麻痺、不随意運動などを呈する。生後早期に発症する先天型と、数ヶ月以降に発症する古典型に大別される。70%はPLP1の重複によるとされ、重複例では古典型を呈することが多い。MRI所見は臨床所見と相関し、先天型ではまったく髄鞘化を認めず白質は広範なT1低信号、T2高信号を呈するが、古典型では内包後脚、視放線、皮質脊髄路などに髄鞘化を認め、同部位はT1高信号、T2低信号を呈する(図1, 7)4)。
2). Pelizaeus-Merzbacher-like disease (PMLD, HDL2, OMIM#608804)
臨床的にはPMD と区別がつかない症状を呈するにもかかわらず,PLP1遺伝子異常を認めない症例をPMLDと呼び,PMDと区別している.一部の症例は、GJC2遺伝子異常を認め PMLD1と診断される。常染色体劣性遺伝であり女性にも発症する。生後早期より眼振を認め,1歳前に運動発達遅滞に気づかれる。その後、錐体路症状,小脳症状,大脳基底核症状が明らかになる。MRI所見は、PMD同様にび漫性白質信号異常を認め、皮質脊髄路は比較的保持(T2低信号)される5, 6)。橋の著明なT2高信号(図7)は特徴的である。拡散強調画像で皮質脊髄路に高信号(拡散能低下)を認めることがある7)。
3). Hypomyelinating leukodystrophy-3 (HLD3, OMOM#260600)
AIMP1遺伝子異常による大脳白質低形成症であり、小頭症、眼振、重度の知的障害、運動発達遅滞を呈する。小頭症や脳波異常の存在から皮質変性症と考えるべきとの意見もある。本邦からの患者報告はない。MRI所見は hypomyelination に合致し、T2強調画像で白質はび漫性に高信号を呈し、T1強調画像では淡い低信号を呈する8)。大脳萎縮、脳梁菲薄化も認められる。MRSでは相対的なNAA低下が報告されている8)。
4). HSP60 chaperon 病 (HLD4, OMIM#612233)
ミトコンドリアシャペロニンHsp60蛋白質の異常による先天性大脳白質形成不全症である。原因遺伝子は、Hsp60遺伝子 (HSPD1) であり本邦からの患者報告はない。眼振、痙性、発達遅滞・退行、約半数にてんかんを認めうる。大脳白質はび漫性にT2高信号を呈し,髄鞘化を全く認めない9)。脳梁の菲薄化と脳室拡大,脳幹と小脳の萎縮を認める。脳幹にT2高信号が認められる。拡散強調画像で皮質脊髄路に高信号(ADC低下)を認める。MRSでは、myo-inositole (mIns)/creatine (Cr)の高値、Choline (Cho)/Crは正常、NAA/Crは正常ないし低値と報告されている9)。
5). Hypomyelination and congenital cataract (HCC, HLD5, OMIM#610532)
大脳白質形成不全と先天性白内障を特徴とする常染色体劣性遺伝疾患であり、FAM126A遺伝子異常による。MRI所見は,hypomyelinationに合致するび漫性のT2高信号(内包後脚も高信号)を呈する10, 11)。脳室周囲、深部白質にはより高度なT2高信号、T1低信号(図6)を認め、同部位は経時的に萎縮が進行する11)。論文では白質の水分含量が増大している可能性が示されている。hypomyelination以外の白質障害(demyelination)も考慮される。皮質下白質に沿ってT1高信号を認めうる。
6). Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC, HLD6).
TUBB4A遺伝子異常に起因する新たな白質変性症であり、運動発達の遅れに引き続き退行、錐体外路症状(ジストニア)、運動失調、痙性麻痺を呈する。知的発達は比較的保たれる。T2強調画像では脳梁、内包後脚にわずかな低信号を認めることがあるが、ほぼ全体に高信号である12, 13)。T1信号は低信号からわずかな高信号まで様々である。加えて、小脳(虫部>半球)、尾状核頭部・被殻、大脳白質の進行性萎縮を呈する(図3)。淡蒼球、視床は保たれる。TUBB4A遺伝子異常患者の一部は、基底核萎縮がなく小脳萎縮のみ認められ、画像所見のみではHDL7, HDL8と鑑別が困難である(図5)13)。
7). Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome, HLD7, OMIM#607694, HLD8, OMIM#614381, HLD11, OMIM#616494)
POLR3A (HDL7), POLR3B (HDL8) 遺伝子異常に起因する白質変性症であり、知的障害、進行性の失調,乏歯・欠歯などの歯牙低形成、下垂体低ゴナドトロピン性性腺機能低下を特徴とする。MRI T2強調画像で大脳白質はび漫性の高信号を呈する14, 15)。併せて小脳萎縮と脳梁菲薄化を認めhypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum (HCAHC) として本邦から報告されていた一群に相当する(図4)14)。POLR3B (HLD8) は、PLOR3A (HLD7) に比べ小脳萎縮が著明とされる16)。近年、POLR1C遺伝子異常も同様の臨床・画像所見を呈することが報告されている17)。
8). Hypomyelinating leukodystrophy-9 (HLD9, OMOM#616140)
RARS遺伝子異常による常染色体劣性遺伝疾患であり、乳児期に運動発達遅滞、知的障害、痙性、眼振を呈する。画像はhypomyelinationに合致し、大脳・小脳の萎縮を認める(図9)18)。MRS ではCholineの低下が報告されている。
9). Hypomyelinating leukodystrophy-10 (HLD10, OMOM#616420)
PYCR遺伝子異常による常染色体劣性遺伝疾患であり、小頭症、運動発達遅滞、知的障害を呈する。T2強調画像で白質は高信号を呈するが、脳梁・内包は低信号である。小頭、白質容量減少、脳梁ひ薄化、脳幹低形成を併せて認める19)。臨床像、画像所見からは皮質変性症の範疇とも考えられる。
10). 18q-症候群 (OMOM#601808)
低身長,精神運動発達遅滞,筋緊張低下,けいれんを呈する.頭部・顔面の形成異常がみられ,小頭症,扁平な顔,眼球離開,心奇形,合指症,難聴,口蓋裂などを来しやすい.生命予後は良好である。大脳白質はT2強調画像にてび漫性に高信号を呈することもあるが、しばしば斑状の高信号を呈する。脳梁、内包後脚はT2低信号を呈することが多い。大脳白質はT1強調画像で等ないし淡い高信号を呈する20)。これらの画像所見をhypomyelinationとは考えにくい。myelin basic protein遺伝子(18q22-23)の欠損により髄鞘形成不全を生じるとされてきたが、病理検討からも髄鞘は正常であり、astrogliosisのためT2高信号を呈するとの報告もある20, 21)。
9). Allan-Herndon-Dudley syndrome (AHDS, OMIM#300523)
X連鎖精神遅滞症候群の1つであり、重度の精神発達遅滞、構音障害、アテトーゼ運動、筋低形成、痙性対麻痺を主症状とし,乳幼児期からMRI検査により髄鞘化の遅れが認められる。大脳白質はT2強調画像にてび漫性に高信号を呈するが、脳梁、内包後脚はT2低信号を呈することが多い22)。T1強調画像では淡い高信号を呈する。経時的に髄鞘化の進行を認める報告が多く、正確にはdelayed myelinationと考えられる。MRSではCho, mIns高値が報告されている22)。
10). Salla病(Salla disease, OMIM#604369)
常染色体劣性遺伝形式をとり,リソゾームへのシアル酸蓄積を特徴とする。原因遺伝子は,SLC17A5遺伝子 (6q14-q15) である.同じ遺伝子変異による infantile sialic acid storage disorder (ISSD, OMIM#269920) は乳児期に致命的であるが,SDは乳児期に発症するが進行は緩徐であり,軽症型ないし成人型とされる。大脳白質はT2強調画像にてび漫性に高信号を呈するが、内包後脚、放線冠はT2低信号(T1高信号)を呈しうる23)。脳梁は菲薄である。MRS検査ではN-acetylaspartate(NAA)の高値を認めるが、遊離シアル酸(N-acethylneuraminic acid; NANA)とNAAを区別できないためとされている。
11). Peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy,
Waardenburg syndrome, and Hirschsprung disease (PCWH, OMIM#609136)
オリゴデンドロサイトの発生異常に加え,Schwann細胞,メラノサイト,腸管ガングリア細胞など神経堤由来細胞の発生異常を本態とする稀な疾患である。臨床的には,中枢神経系の髄鞘形成不全に加え,末梢神経系では脱髄型ニューロパチーを呈し,さらにWaardenburg症候群,Hirschsprung病を合わせた4症候群を合併する。原因遺伝子はSOX10で,常染色体優性遺伝形式をとるが,多くは突然変異による弧発例である。T2強調画像にて全大脳白質はび漫性高信号を呈し、大脳萎縮を伴う24)。一方,軽症例では経時的な髄鞘形成を認め髄鞘化遅延(delayed myelination)の所見を呈する25)。
12). その他
画像上、hypomyelination pattern を呈しうる疾患として
Hypomyelination with brain stem and spinal cord involvement and leg spasticity (HBSL, DARS遺伝子異常), Trichothiodystrophy with hypersensitivity to sunlight (Tay syndrome), Fucosidosis(図8), Serine synthesis defects, Oculodentodigital dysplasia (ODDD), Galactosemia, Cockayne syndrome, early onset neuronal degenerative disorders (early onset GM1, GM2 gangliosidosis, infantile neuronal ceroid lipofuscinosis, Alpers syndrome), mucolipidosis type II (I-cell disease), type IV, MCPH10 (ZNF335遺伝子異常) 26)(図10)
が列挙される。
文献()内、エビデンスレベル
- Barkovich AJ, Deon S. Hypomyelinating disorders: an MRI approach. Neurobiol Dis 2016; 87: 50-58.(6)
- Schiffmann R, van der Knaap MS. An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009; 72: 750-759.(6)
- Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010; 133; 2971-2982. (4b)
- Takanashi J, Sugita K, Tanabe Y, et al. MR-revealed myelination in the cerebral corticospinal tract as a marker for Pelizaeus-Merzbacher's disease with proteolipid protein gene duplication. AJNR Am J Neuroradiol 1999; 20: 1822-1828. (5)
- Uhlenberg B, Schuelke M, Ruschendorf F, et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004; 75:251–260. (5)
- Abrams CK, Scherer SS. Gap junctions in inherited human disorders of the central nervous system. Biochem Biophys Acta 2012; 1818; 2030-2047.(6)
- Biancheri R, Rosano C, Denegri L, et al. Expanded spectrum of Pelizaeus–Merzbacher-like disease: literature revision and description of a novel GJC2 mutation in an unusually severe form. Eur J Hum Genet 2013; 21: 34-39.(5)
- Feinstein M, Markus B, Noyman I, et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am J Hum Genet 2010; 87: 820-828. (5)
- Magen D, Georgopoulos C, Bross P, et al. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet 2008; 83: 30-42. (5)
- Zara F, Biancheri R, Bruno C, et al. Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat Genet 2006; 38: 1111-3. (5)
- Biancheri R, Zara F, Rossi A, et al. Hypomyelination and congenital cataract. Broadening the clinical phenotype. Arch Neurol 2011; 68: 1191-1194. (5)
- Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 2013; 92: 767-73. (5)
- Miyatake S, Osaka H, Shiina M, et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology 2014; 82: 2230-7. (5)
- Sasaki M, Takanashi J, Tada H, et al: Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev 2009; 31: 582-587. (5)
- Saitsu H, Osaka H,Sasaki M, et al: Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal recessive hypomyelinating leukoencephalopathy. Am J Hum Genet 2011; 89: 644-651. (5)
- Takanashi J, Osaka H, Saitsu H, et al. Different patterns of hypomyelination and cerebellar abnormality between POLR3A and POLR3B mutations. Brain Dev 2014; 36: 259-263. (5)
- Thiffault I, Wolf NI, Forget D, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun 2015; 6: 7623. doi: 10.1038/ncomms8623 (5)
- Wolf NI, Salomons GS, Rodenburg RJ, et al. Mutations in RARS cause hypomyelination. Ann Neurol 2014; 76: 134-139. (5)
- Nakayama T, Al-Maawali A, El-Quessny M, et al. Mutations in PYCR2, encoding pyrroline-5-carboxylate reductase 2, cause microcephaly and hypomyelination. Am J Hum genet 2015 96: 709-719. (5)
- Tada H, Takanashi J. MR spectroscopy in 18q- syndrome suggesting other than hypomyelination. Brain Dev 2014; 36: 57-60. (5)
- Tanaka R, Iwasaki N, Hayashi M, et al. Abnormal brain MRI signal in 18q- syndrome
not due to dysmyelination. Brain Dev 2012; 34: 234–237. (5) - Gika AD, Siddiqui A, Hulse AJ, White matter abnormalities and dystonic motor disorder associated with mutations in the SLC16A2 gene. Dev Med Child Neurol 2010; 52: 475-482. (5)
- Sonninen P, Autti T, Varho T, et al. Brain involvement in Salla disease. Am J Neuroradiol 1999; 20: 433-443. (5)
- Inoue K, Tanabe Y, Lupski JR. Myelin deficiencies in both the central and the peripheral nervous system associated with a SOX10 mutation. Ann Neurol 1999; 46: 313-318. (5)
- Verheij JB, Sival DA, van der Hoeven JH, et al. Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: a case report and review of the literature. Eur J Paediatr Neurol. 2006; 10:11-17. (5)
- Sato R, Takanashi J, Tsuyusaki Y, Kato M, Saitsu H, Komiyama O, Takahashi T. Association between invisible basal ganglia and ZNF335 mutations: a case report. Pediatrics 2016; e20160897, DOI: 10.1542/peds.2016-0897. (5)
Pub-med 検索式
- hypomyelination[All Fields] AND ("magnetic resonance imaging"[MeSH Terms] OR ("magnetic"[All Fields] AND "resonance"[All Fields] AND "imaging"[All Fields]) OR "magnetic resonance imaging"[All Fields] OR "mri"[All Fields])
251件









