








Guidelines on Hereditary Leukodystrophies
CQ2 What sort of imaging is used to diagnose cerebral leukodystrophy?
The use of magnetic resonance imaging to diagnose cerebral leukodystrophy is strongly recommended. (Recommendation grade A)
The characteristic imaging finding is overall faint hyperintensity of the white matter on T2-weighted imaging. Depending on the disorder, additional specific imaging signs may appear and provide evidence for the diagnosis.
Explanation
Cerebral leukodystrophies are a group of diseases involving impaired or delayed myelin formation, and the magnetic resonance imaging (MRI) scans of affected patients resemble those of newborn babies with immature myelination. Compared with cortical tissue, the white matter appears hypointense on T1-weighted imaging and hyperintense on T2-weighted imaging. On T2-weighted imaging, the white matter appears faintly hyperintense overall, with hypointensities reflecting myelination only apparent at the locations where myelination first occurs (such as the posterior limb of the internal capsule, optic radiation, and corticospinal tract) (Figure 1).1–3) In the myelination-associated changes seen in MRI signal intensity, hyperintensity on T1-weighted imaging is known to appear before hypointensity on T2-weighted imaging. Various patterns may be evident on T1-weighted imaging depending on the degree of myelin formation: the white matter may be mildly hypointense overall, there may be faint hyperintensities only at the locations where the myelination first occurs, or widespread faint hyperintensity may be evident. For a definitive diagnosis of cerebral leukodystrophy on the basis of MRI findings, abnormal white matter signal intensity must remain unchanged for at least 6 months, and an MRI evaluation must be performed at least once after the patient turns 1 year old. If MRI reveals signs of severe hypomyelination after 2 years of age, cerebral leukodystrophy is highly likely.2, 3) Figure 2 shows a flow chart of the imaging diagnosis of the various conditions classified under the general concept of cerebral leukodystrophy.
These are summarized below with an explanation of the imaging findings that are characteristic of each disease.
Flow chart of the imaging diagnosis of cerebral leukodystrophy (Figure 2)
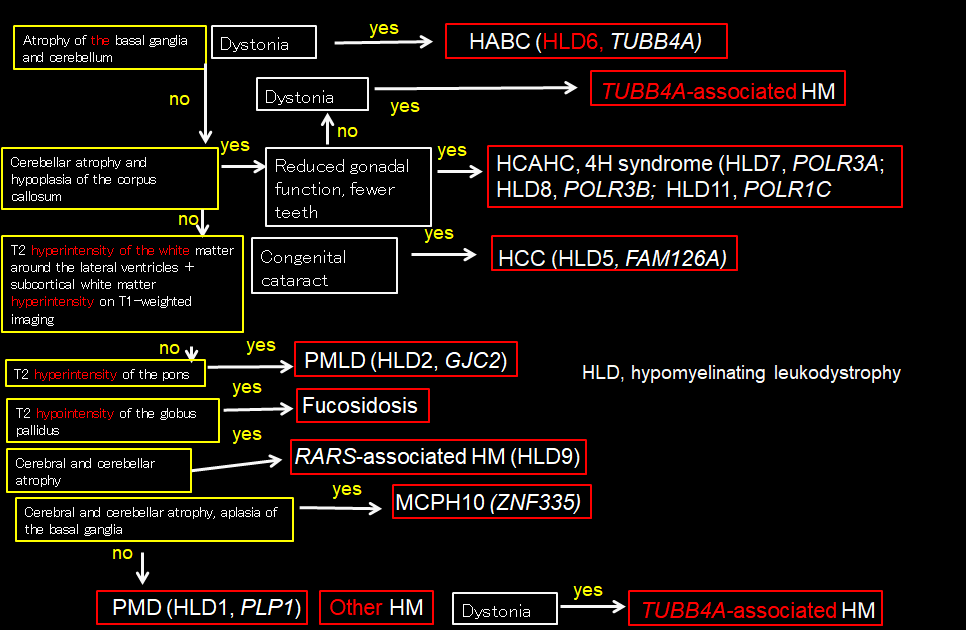
This flow chart summarizes the diagnostic flow chart for patients with overall faint hyperintensity of the white matter on T2-weighted imaging suggestive of cerebral leukodystrophy. The yellow boxes indicate imaging findings, the white boxes clinical symptoms, and the red boxes diagnoses.
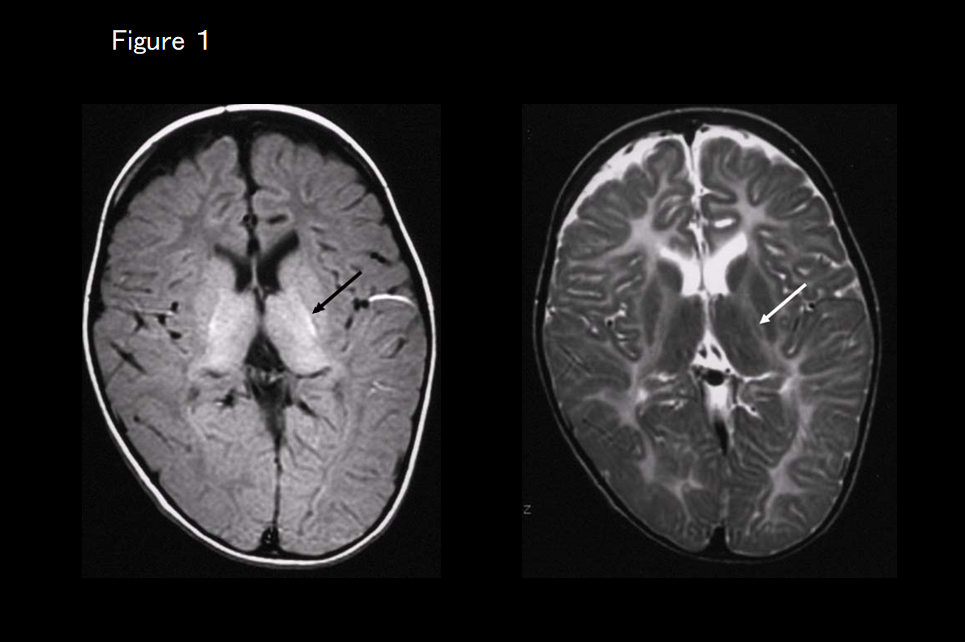
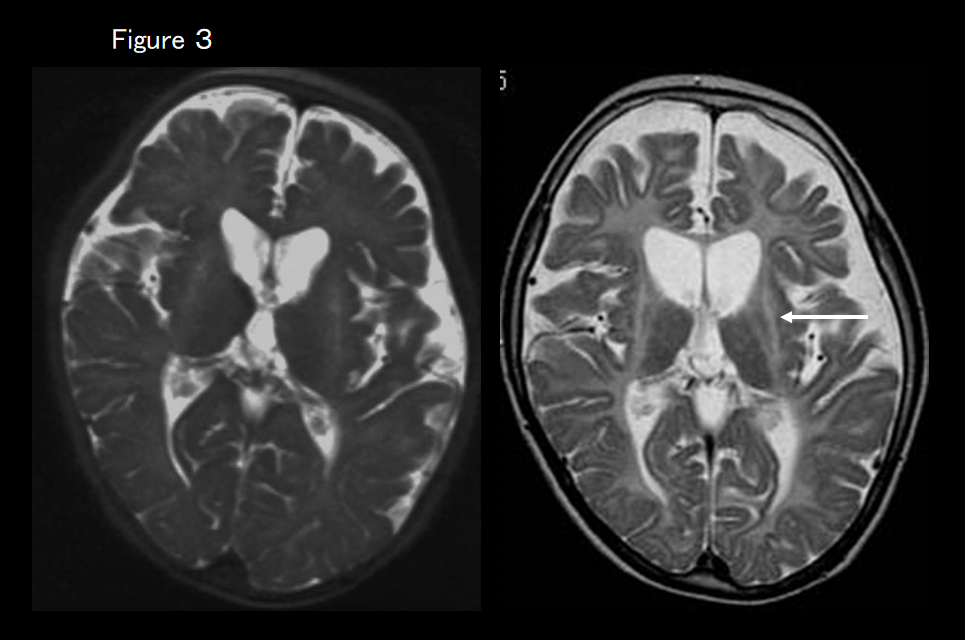
If MRI shows atrophy of the basal ganglia and cerebellum in addition to cerebral leukodystrophy (Figure 3), the first suspected diagnosis is hypomyelination with atrophy of the basal ganglia and cerebellum. In most cases, its clinical symptoms include extrapyramidal symptoms, of which dystonia is the most important, and screening for the causative TUBB4A gene is performed.
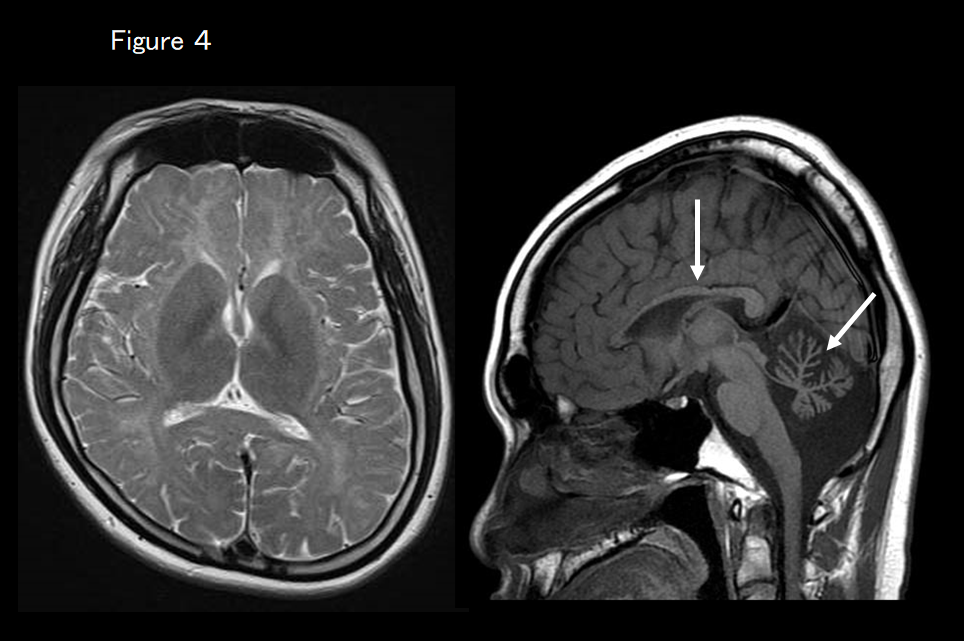
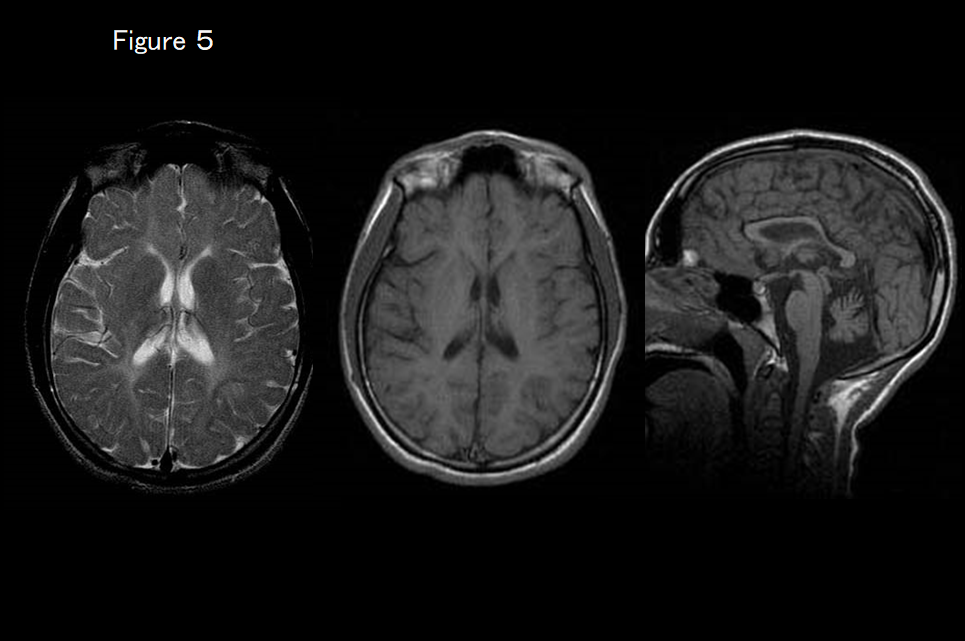
If MRI shows that the basal ganglia are normal but atrophy of the cerebellum and thinning of the corpus callosum (hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum) are present (Figure 4), peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia is suspected. It is essential to check gonadal function and tooth number as well as screen for the causative genes POLR3A, 3B, and 1C. If MRI findings indicate HCAHC and the basal ganglia are normal but dystonia is clinically present (Figure 5), TUBB4A-associated hypomyelinating disorder is suspected.
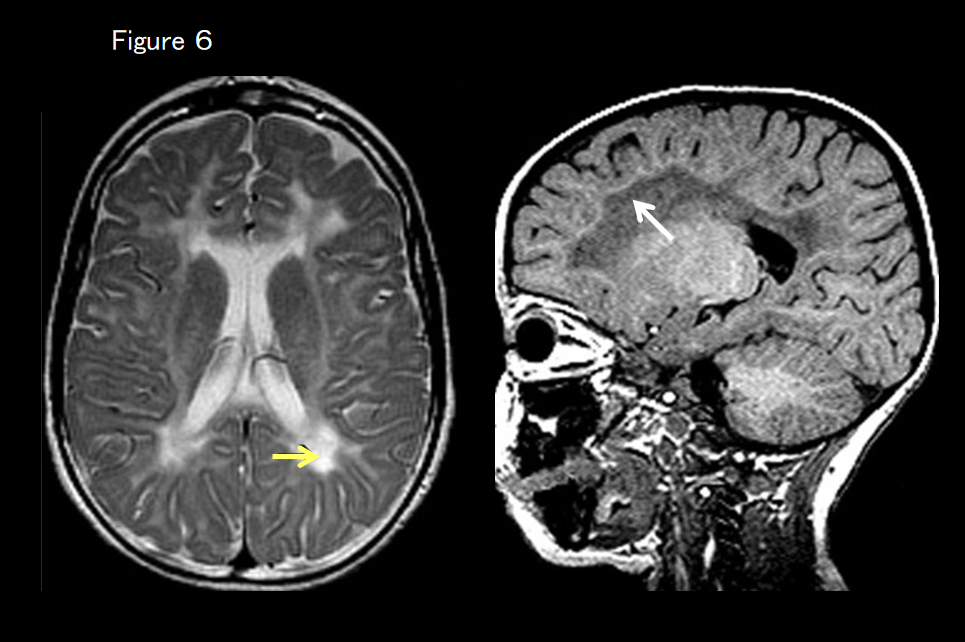
If there are no basal ganglia or cerebellar findings but there are marked T2-weighted hyperintensities around the lateral ventricles (more strongly hyperintense on T2-weighted imaging than the hyperintensity apparent in cerebral hypomyelination), and the subcortical white matter is hyperintense on T1-weighted imaging (Figure 6), hypomyelination and congenital cataract are suspected. Ophthalmic screening (for cataract) is then essential, as is screening for the FAM126A gene.
If none of the above applies, the following imaging findings are examined.
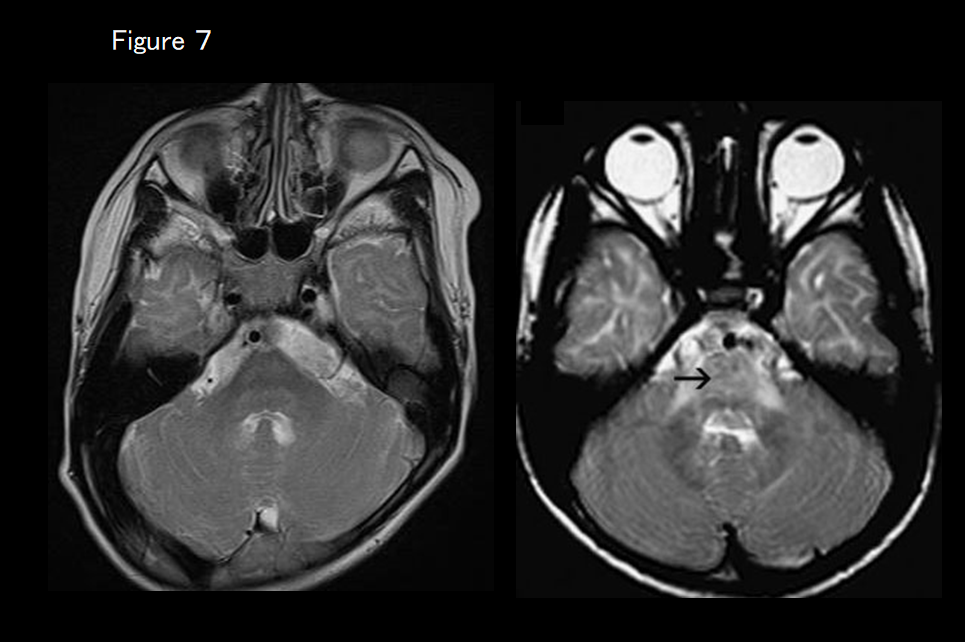
If there is marked T2 hyperintensity of the pons (Figure 7), Pelizaeus-Merzbacher–like disease (PMLD; HLD2, GJC2 mutation) is suspected.
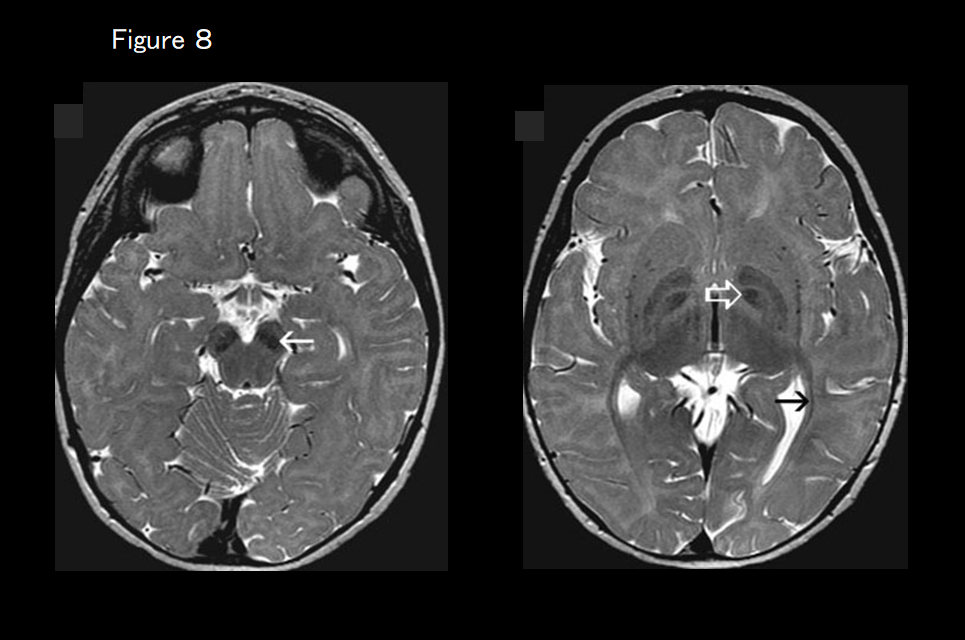
If marked T2 hypointensity of the globus pallidus is present (Figure 8), fucosidosis is suspected.
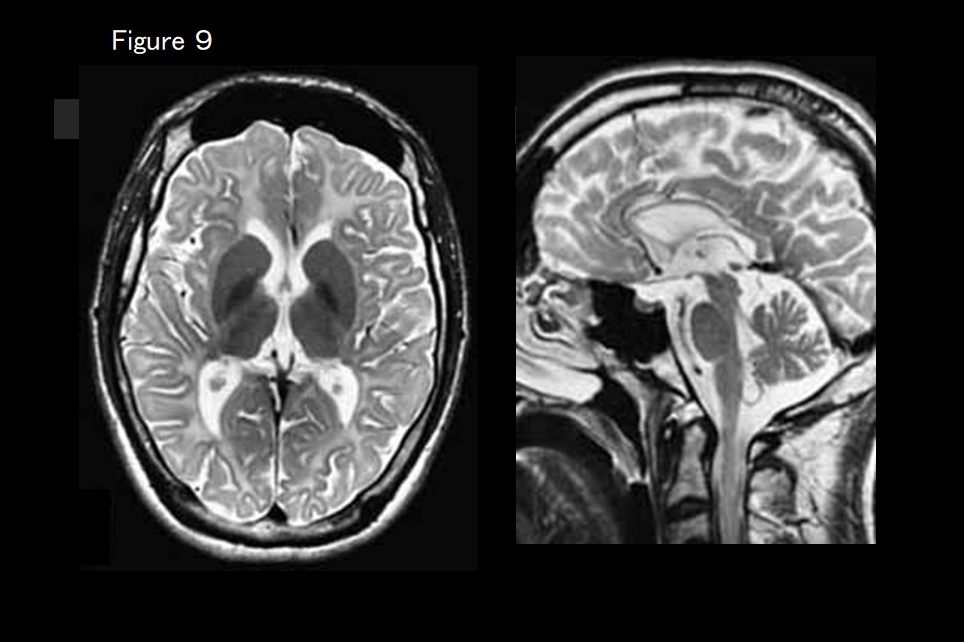
If cerebral and cerebellar atrophy are present (Figure 9), hypomyelinating leukodystrophy-9 (RARS mutation) is suspected.
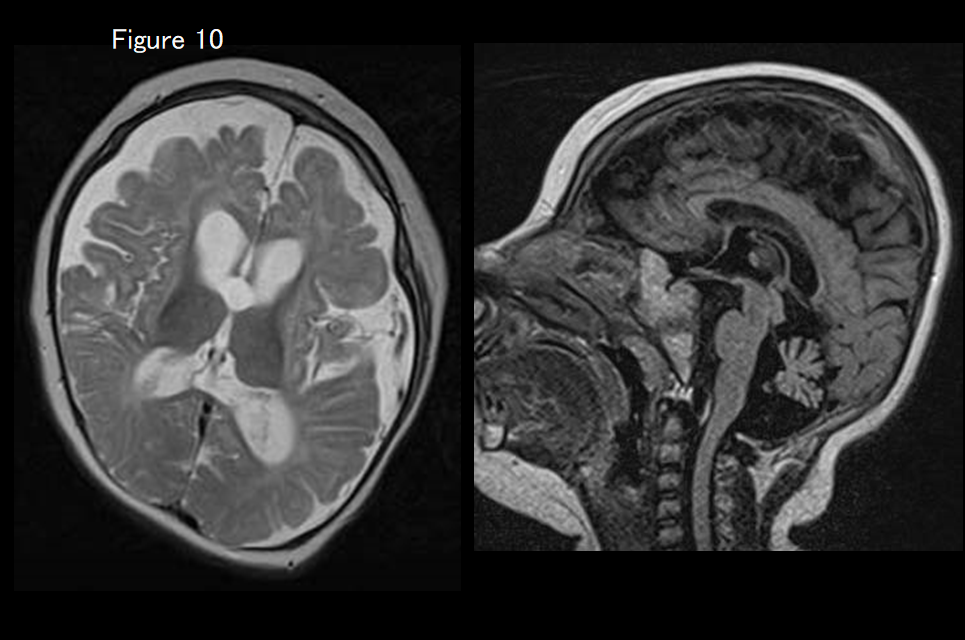
If cerebral and cerebellar atrophy, hypoplasia of the brainstem, and aplasia of the basal ganglia are evident (Figure 10), MCPH10 (ZNF335 mutation) is suspected.
If patients exhibit findings that are inconsistent with any of the above, the diagnostic process for other types of cerebral leukodystrophy, including Pelizaeus-Merzbacher disease (PMD; HLD1), is used. If cerebral leukodystrophy is the only imaging finding observed but dystonia is also clinically present, then TUBB4A-associated hypomyelinating disorder is suspected.
Individual diseases
1). Pelizaeus-Merzbacher disease (PMD, HLD1, OMIM#312080)
PMD is a sex-linked autosomal recessive inherited disease caused by an abnormality in the gene that encodes proteolipid protein 1 (PLP1), the main constituent of myelin, with other symptoms including congenital nystagmus, head bobbing, spastic palsy, and involuntary movement. It is broadly classified into two forms: the connatal form, which appears soon after birth; and the classical form, which appears after several months. In 70% of cases, PMD is caused by PLP1 gene duplication; in most of these patients, it takes the classical form. MRI findings are correlated with the clinical symptoms: In the connatal form, no myelination at all is present, with widespread T1 hypointensity and T2 hyperintensity in the white matter; in the classical form, myelination of areas such as the posterior limb of the internal capsule, optic radiation, and corticospinal tract has occurred and these locations appear hyperintense on T1-weighted imaging and hypointense on T2-weighted imaging (Figures 1 and 7).4)
2). Pelizaeus-Merzbacher-like disease (PMLD, HDL2, OMIM#608804)
Patients with PMLD have symptoms that are clinically indistinguishable from those of PMD but without any PLP1 gene abnormalities. In some patients, an abnormality of the GJC2 gene is present; these cases are diagnosed as PMLD1. PMLD is an autosomal recessive inherited disease that is also seen in females. Nystagmus appears soon after birth, and delayed motor development is noticeable before 1 year of age. Pyramidal, cerebellar, and corticobasal ganglia symptoms subsequently become apparent. MRI findings comprise the same diffuse white-matter abnormalities seen in PMD, but the corticospinal tract is comparatively spared (hypointense on T2-weighted imaging).5, 6) Marked T2 hyperintensity of the pons (Figure 7) is characteristic. The corticospinal tract may appear hyperintense (diminished diffusion capacity) on diffusion-weighted imaging.7)
3). Hypomyelinating leukodystrophy-3 (HLD3, OMOM#260600)
This form of cerebral hypomyelination is caused by an AIMP1 mutation causing microcephaly, nystagmus, severe intellectual disability, and delayed motor development. Some authors believe that HLD3 should be considered a cortical degenerative disease due to the presence of microcephaly and brainwave abnormalities. No cases have been reported in Japan. MRI findings are consistent with hypomyelination, showing diffuse hyperintensity of the white matter on T2-weighted imaging and mild hypointensity on T1-weighted imaging. 8) Cerebral atrophy and thinning of the corpus callosum are also present. Relatively low N-acetylaspartate (NAA) on magnetic resonance spectroscopy (MRS) has also been reported.8)
4). HSP60 chaperon 病 (HLD4, OMIM#612233)
This form of congenital cerebral leukodystrophy is caused by an abnormality in the mitochondrial chaperone protein Hsp60. The causative gene is Hsp60 (HSPD1); no cases have been reported from Japan. Nystagmus, spasticity, developmental delay, or regression may also be present, as is epilepsy in approximately 50% of cases. The cerebral white matter exhibits diffuse T2 hyperintensity with no myelination at all.9) Thinning of the corpus callosum, ventricular enlargement, and atrophy of the brainstem and cerebellum are also present. The brainstem is hyperintense on T2-weighted imaging. The corticospinal tract is hyperintense (low apparent diffusion coefficient) on diffusion-weighted imaging. High myo-inositol (mIns)/creatine (Cr), normal choline (Cho)/Cr, and normal or low NAA/Cr values on MRS have also been reported.9)
5). Hypomyelination and congenital cataract (HCC, HLD5, OMIM#610532)
This is an autosomal recessive inherited disease characterized by cerebral hypomyelination and congenital cataract caused by a FAM126A mutation. MRI shows diffuse hyperintensity on T2-weighted imaging (including hyperintensity of the posterior limb of the internal capsule) consistent with hypomyelination.10, 11) The periventricular and deep white matter are even more hyperintense on T2-weigted imaging and hypointense on T1-weighted imaging (Figure 6), and these areas become increasingly atrophied over time.11) Studies have suggested that the water content of the white matter may increase. In addition to hypomyelination, other white-matter disorders (demyelination) are considered. T1-weighted hyperintensity along the subcortical white matter may also be seen.
6). Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC, HLD6).
This is a newly identified white-matter degenerative disease caused by a TUBB4A mutation that causes the delay and subsequent regression of motor development, extrapyramidal symptoms (dystonia), motor ataxia, and spastic palsy. Intellectual development is comparatively maintained. Although very slight hypointensity of the corpus callosum and the posterior limb of the internal capsule may be evident on T2-weighted imaging, almost all of the white matter is hyperintense.12, 13) The signal on T1-weighted imaging varies from hypointense to very slightly hyperintense. Progressive atrophy of the cerebellum (vermis > hemisphere), caudate nucleus and putamen, and cerebral white matter is also evident (Figure 3). The globus pallidus and thalamus are spared. In some patients with a TUBB4A mutation, there is no atrophy of the basal ganglion and only the cerebellum is atrophied, making it difficult to distinguish from HLD7 and HLD8 based on imaging findings alone (Figure 5). 13)
7). Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia (4H syndrome, HLD7, OMIM#607694, HLD8, OMIM#614381, HLD11, OMIM#616494)
This is a white-matter degenerative disease caused by a POLR3A (HLD7) or POLR3B (HLD8) mutation characterized by intellectual disability, progressive ataxia, hypodontia (e.g., oligodontia or missing teeth), and pituitary hypogonadotropic hypogonadism. MRI reveals diffuse hyperintensity of the cerebral white matter on T2-weighted imaging.14, 15) Cerebellar atrophy and thinning of the corpus callosum are also present, corresponding to the cases reported from Japan of hypomyelination with cerebellar atrophy and HCAHC (Figure 4).14) Cerebral atrophy is more severe in patients with abnormal POLR3B (HLD8) than those with abnormal PLOR3A (HLD7).16) It was recently reported that similar clinical symptoms and imaging findings may also be present in patients with POLR1C mutation.17)
8). Hypomyelinating leukodystrophy-9 (HLD9, OMOM#616140)
This is an autosomal recessive inherited disease caused by a RARS mutation that causes delayed motor development, intellectual disability, spasticity, and nystagmus in infancy. The imaging presentation is consistent with hypomyelination, and cerebral and cerebellar atrophy are present (Figure 9).18) Low Cho on MRS has been reported.
9). Hypomyelinating leukodystrophy-10 (HLD10, OMOM#616420)
This is an autosomal recessive inherited disease caused by a PYCR mutation that causes microcephaly, delayed motor development, and intellectual disability. The white matter is hyperintense on T2-weighted imaging, whereas the corpus callosum and internal capsule are hypointense. Microcephaly, decreased white matter volume, thinning of the corpus callosum, and hypoplasia of the brainstem are also present.19) Because of its clinical presentation and imaging findings, it is also considered to fall within the taxonomy of cortical degenerative diseases.
10). Chromosome 18q deletion syndrome (OMOM#601808)
This causes short stature, delayed psychomotor development, hypotonia, and seizures. Cranial and facial dysplasia are evident, and abnormalities including microcephaly, facial flattening, hypertelorism, cardiac malformation, syndactyly, hearing impairment, and cleft lip are common. The survival prognosis is good. There may be diffuse hyperintensity of the white matter on T2-weighted imaging, but patches of hyperintensity are usually present. The corpus callosum and the posterior limb of the internal capsule are often hypointense on T2-weighted imaging. On T1-weighted imaging, the cerebral white matter is either isointense or faintly hyperintense.20) These imaging findings are not indicative of hypomyelination; rather, they are believed to stem from dysmyelination due to deletion of the gene that encodes the myelin basic protein (18q22–23), but some studies reported that pathological testing revealed normal myelin, with the T2-weighted hyperintensities actually caused by astrogliosis.20, 21)
9). Allan-Herndon-Dudley syndrome (AHDS, OMIM#300523)
The main symptoms of this X-linked mental retardation syndrome are severely delayed mental development, dysarthria, athetoid movements, muscle hypoplasia, and spastic paraplegia, and delayed myelination is evident on MRI from infancy. There is diffuse hyperintensity of the white matter on T2-weighted imaging, and the corpus callosum and the posterior limb of the internal capsule are often hypointense.22) On T1-weighted imaging, the white matter is faintly hyperintense. In many cases, myelination increases over time and should accurately be described as delayed myelination. High Cho and mIns levels on MRS have been reported.22)
10). Salla disease (OMIM #604369)
This is an autosomal recessive inherited disease characterized by the accumulation of sialic acid in lysosomes. The causative gene is SLC17A5 (6q14-q15). Infantile sialic acid storage disorder (OMIM #269920), which is caused by another mutation in the same gene, is fatal in infancy; although the first symptoms of Salla disease emerge in infancy, its progression is gradual and it is considered a mild adult disease. There is diffuse hyperintensity of the white matter on T2-weighted imaging, but the corpus callosum and the posterior limb of the internal capsule may be hypointense on T2-weighted imaging (hyperintense on T1-weighted imaging).23) The corpus callosum is thin. NAA is high on MRS, but this is because this investigation cannot distinguish between N-acethylneuraminic acid and NAA.
11). Peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy,
Waardenburg syndrome, and Hirschsprung disease (PCWH, OMIM#609136)
This is a rare condition characterized by abnormal oligosaccharide generation and the abnormal generation of Schwann cells, melanocytes, intestinal ganglion cells, and other neural crest–derived cells. Clinically, it comprises a combination of four different syndromes: dysmyelination of the central nervous system, demyelinating neuropathy of the peripheral nervous system, Waardenburg syndrome, and Hirschsprung disease. It is an autosomal dominant inherited condition caused by mutation of the SOX10 gene, and most cases are sporadic mutations. Diffuse hyperintensity of the white matter is visible on T2-weighted imaging, and cerebral atrophy is present.24) However, mild cases exhibit signs of delayed myelination with myelin formation occurring over time. 25)
12). Others
Other diseases that may exhibit a hypomyelination pattern on diagnostic imaging include hypomyelination with brain stem and spinal cord involvement and leg spasticity (DARS mutation), trichothiodystrophy with hypersensitivity to sunlight (Tay syndrome), fucosidosis (Figure 8), serine synthesis defects, oculodentodigital dysplasia, galactosemia, Cockayne syndrome, early-onset neuronal degenerative disorders (early-onset GM1, GM2 gangliosidosis, infantile neuronal ceroid lipofuscinosis, Alpers syndrome), mucolipidosis type II (I-cell disease) or type IV, and MCPH10 (ZNF335 mutation)(Figure 10).26)
References (evidence level in parentheses)
- Barkovich AJ, Deon S. Hypomyelinating disorders: an MRI approach. Neurobiol Dis 2016; 87: 50-58.(6)
- Schiffmann R, van der Knaap MS. An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009; 72: 750-759.(6)
- Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010; 133; 2971-2982. (4b)
- Takanashi J, Sugita K, Tanabe Y, et al. MR-revealed myelination in the cerebral corticospinal tract as a marker for Pelizaeus-Merzbacher's disease with proteolipid protein gene duplication. AJNR Am J Neuroradiol 1999; 20: 1822-1828. (5)
- Uhlenberg B, Schuelke M, Ruschendorf F, et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004; 75:251–260. (5)
- Abrams CK, Scherer SS. Gap junctions in inherited human disorders of the central nervous system. Biochem Biophys Acta 2012; 1818; 2030-2047.(6)
- Biancheri R, Rosano C, Denegri L, et al. Expanded spectrum of Pelizaeus–Merzbacher-like disease: literature revision and description of a novel GJC2 mutation in an unusually severe form. Eur J Hum Genet 2013; 21: 34-39.(5)
- Feinstein M, Markus B, Noyman I, et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am J Hum Genet 2010; 87: 820-828. (5)
- Magen D, Georgopoulos C, Bross P, et al. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet 2008; 83: 30-42. (5)
- Zara F, Biancheri R, Bruno C, et al. Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat Genet 2006; 38: 1111-3. (5)
- Biancheri R, Zara F, Rossi A, et al. Hypomyelination and congenital cataract. Broadening the clinical phenotype. Arch Neurol 2011; 68: 1191-1194. (5)
- Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 2013; 92: 767-73. (5)
- Miyatake S, Osaka H, Shiina M, et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology 2014; 82: 2230-7. (5)
- Sasaki M, Takanashi J, Tada H, et al: Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev 2009; 31: 582-587. (5)
- Saitsu H, Osaka H,Sasaki M, et al: Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal recessive hypomyelinating leukoencephalopathy. Am J Hum Genet 2011; 89: 644-651. (5)
- Takanashi J, Osaka H, Saitsu H, et al. Different patterns of hypomyelination and cerebellar abnormality between POLR3A and POLR3B mutations. Brain Dev 2014; 36: 259-263. (5)
- Thiffault I, Wolf NI, Forget D, et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat Commun 2015; 6: 7623. doi: 10.1038/ncomms8623 (5)
- Wolf NI, Salomons GS, Rodenburg RJ, et al. Mutations in RARS cause hypomyelination. Ann Neurol 2014; 76: 134-139. (5)
- Nakayama T, Al-Maawali A, El-Quessny M, et al. Mutations in PYCR2, encoding pyrroline-5-carboxylate reductase 2, cause microcephaly and hypomyelination. Am J Hum genet 2015 96: 709-719. (5)
- Tada H, Takanashi J. MR spectroscopy in 18q- syndrome suggesting other than hypomyelination. Brain Dev 2014; 36: 57-60. (5)
- Tanaka R, Iwasaki N, Hayashi M, et al. Abnormal brain MRI signal in 18q- syndrome
not due to dysmyelination. Brain Dev 2012; 34: 234–237. (5) - Gika AD, Siddiqui A, Hulse AJ, White matter abnormalities and dystonic motor disorder associated with mutations in the SLC16A2 gene. Dev Med Child Neurol 2010; 52: 475-482. (5)
- Sonninen P, Autti T, Varho T, et al. Brain involvement in Salla disease. Am J Neuroradiol 1999; 20: 433-443. (5)
- Inoue K, Tanabe Y, Lupski JR. Myelin deficiencies in both the central and the peripheral nervous system associated with a SOX10 mutation. Ann Neurol 1999; 46: 313-318. (5)
- Verheij JB, Sival DA, van der Hoeven JH, et al. Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: a case report and review of the literature. Eur J Paediatr Neurol. 2006; 10:11-17. (5)
- Sato R, Takanashi J, Tsuyusaki Y, Kato M, Saitsu H, Komiyama O, Takahashi T. Association between invisible basal ganglia and ZNF335 mutations: a case report. Pediatrics 2016; e20160897, DOI: 10.1542/peds.2016-0897. (5)
Pubmed Search Format
- hypomyelination[All Fields] AND ("magnetic resonance imaging"[MeSH Terms] OR ("magnetic"[All Fields] AND "resonance"[All Fields] AND "imaging"[All Fields]) OR "magnetic resonance imaging"[All Fields] OR "mri"[All Fields])
251 results