
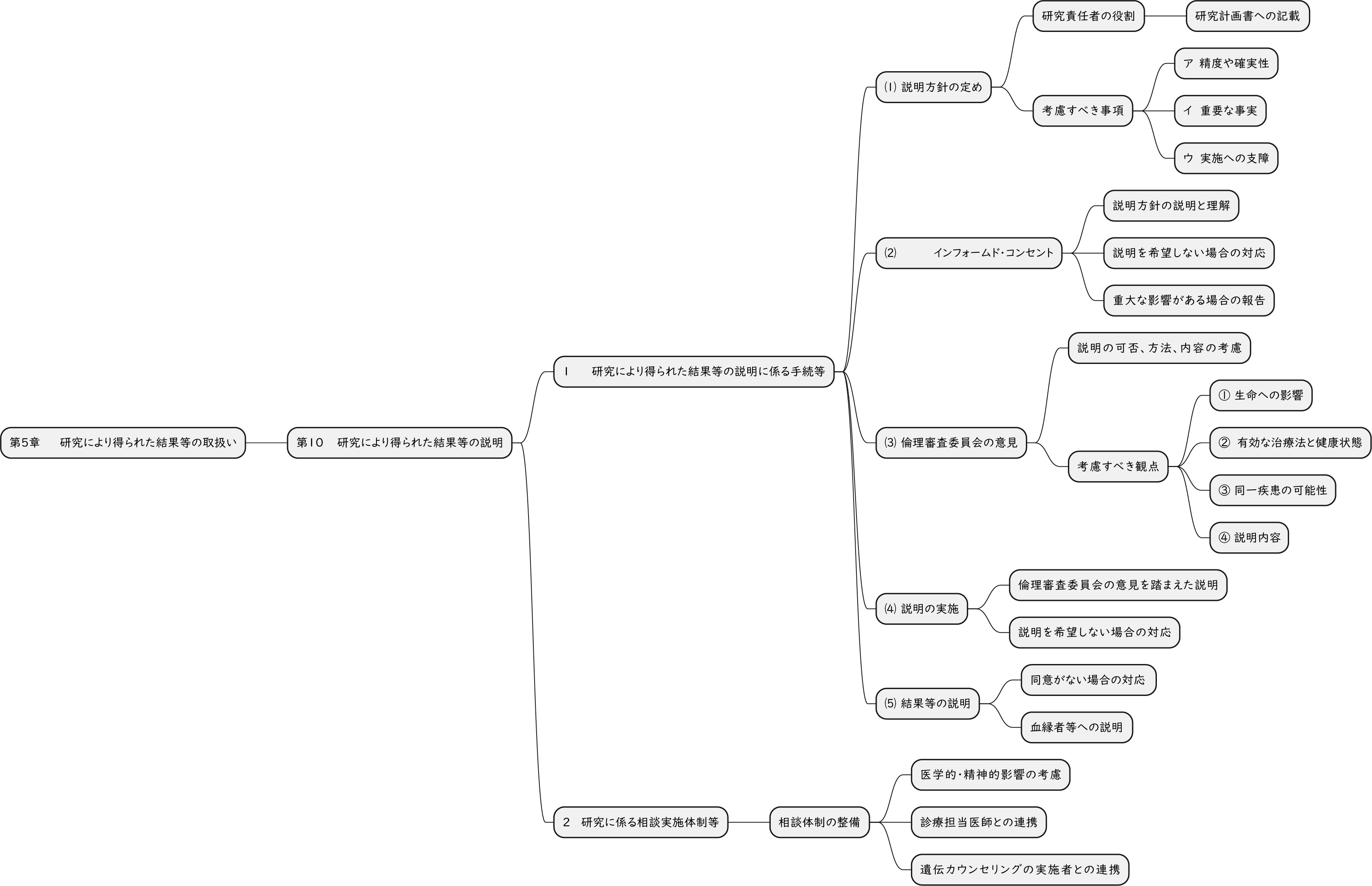
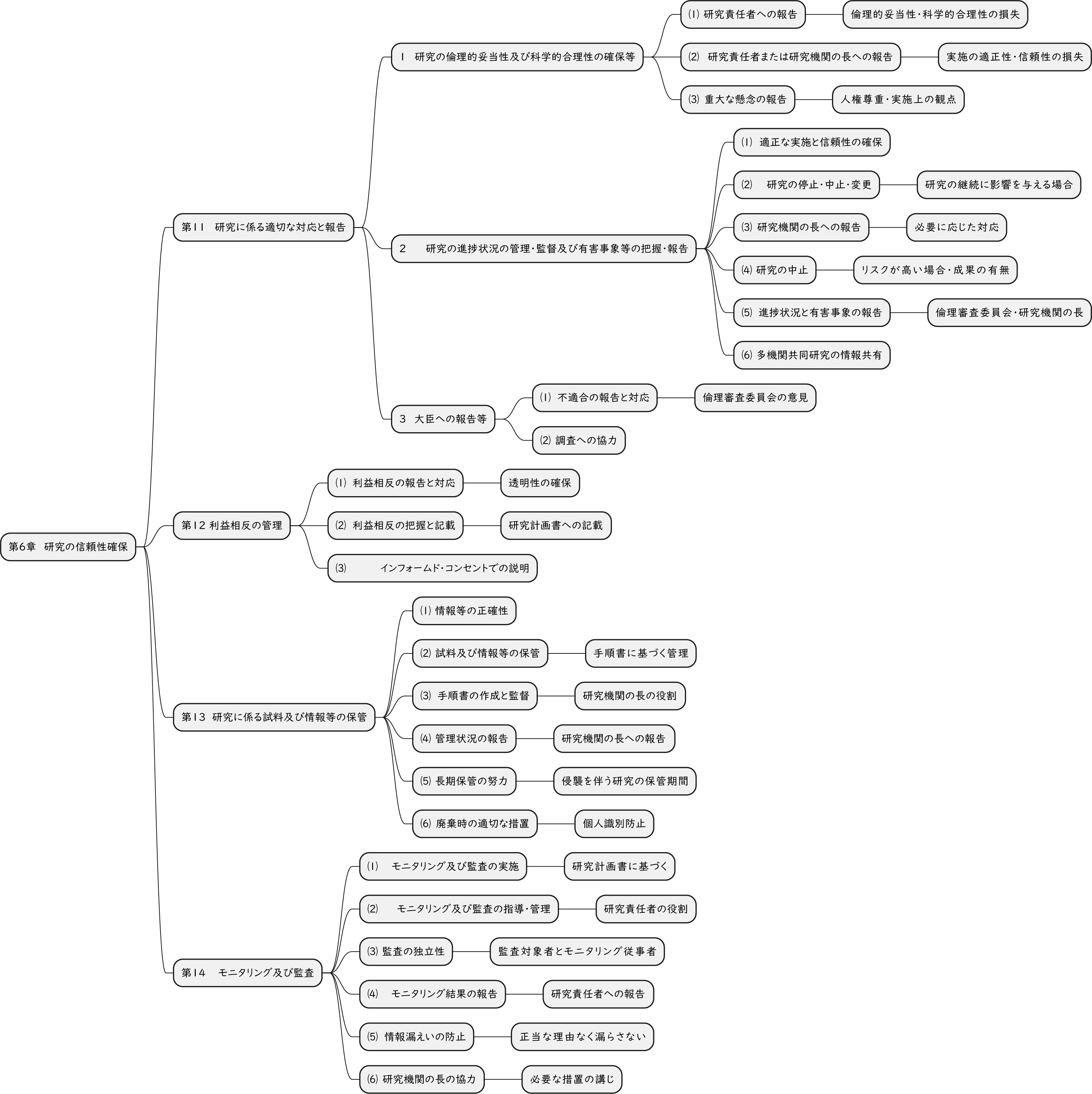
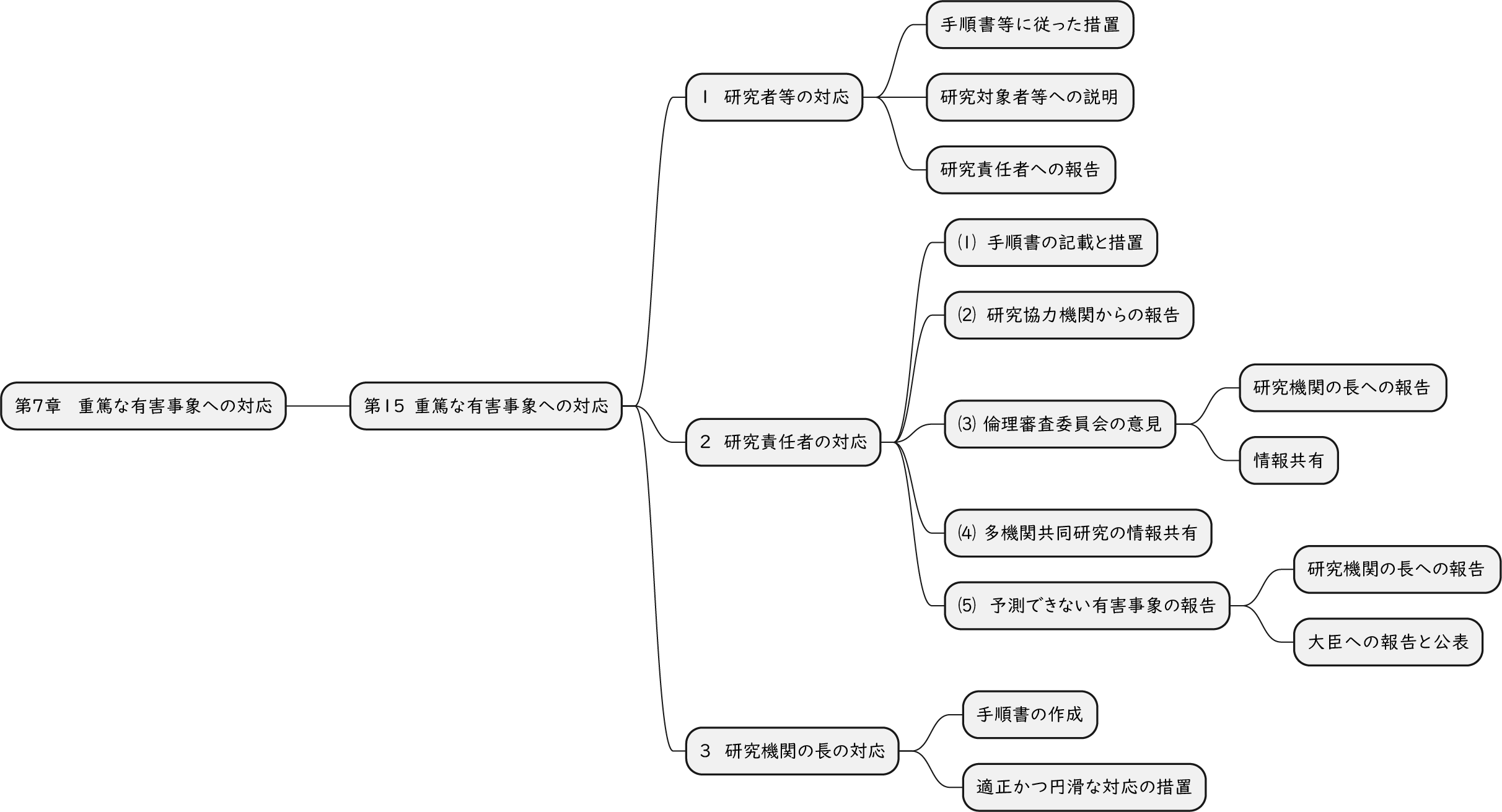
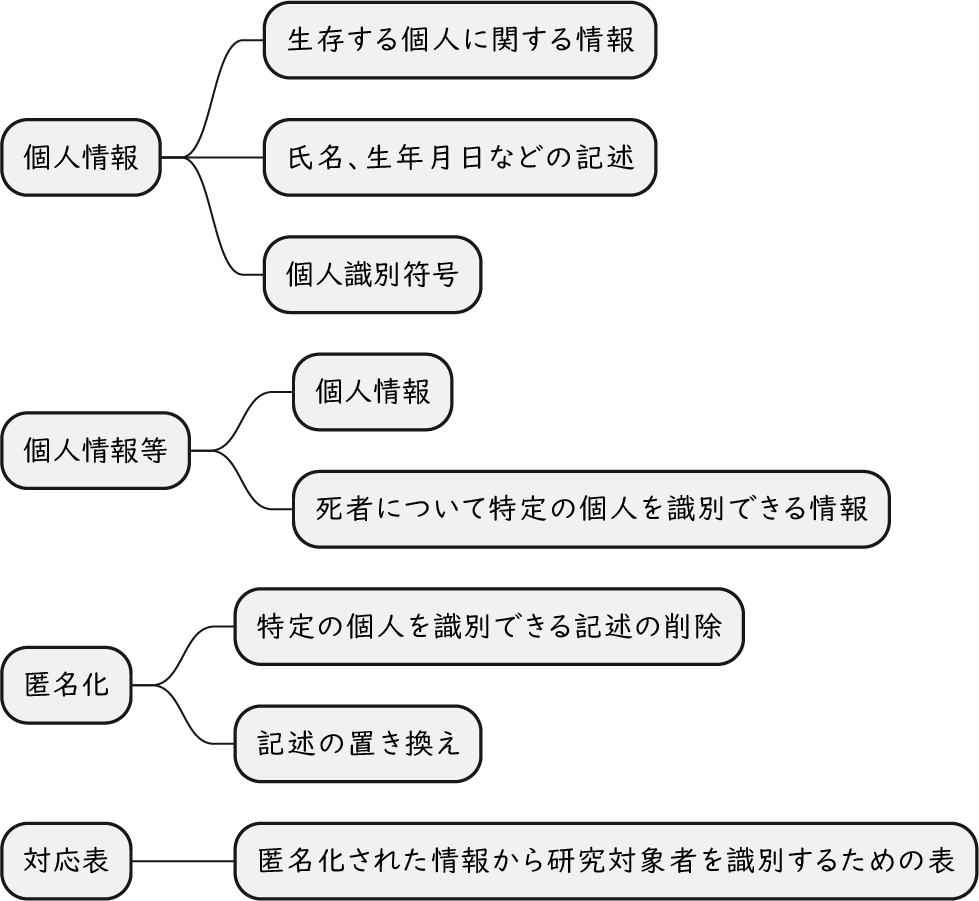
ざっくり何について書かれているのかを把握しやすいようなマインドマップにまとめてみました。情報の抽出にはCopilotを使用しました。ところどころおかしなところがあるのは把握しています。気が向いたら修正したいとは思っていますが、とりあえず、そのまま掲載しています。どこの項に知りたいことが書かれているのかを探す向きにはこういうまとめも有用だろうと思います。実務に重要なポイントは元の文章を確認する必要があります。
もとになったのは平成29年版
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Q&A 長いので2つのパートに分割しました


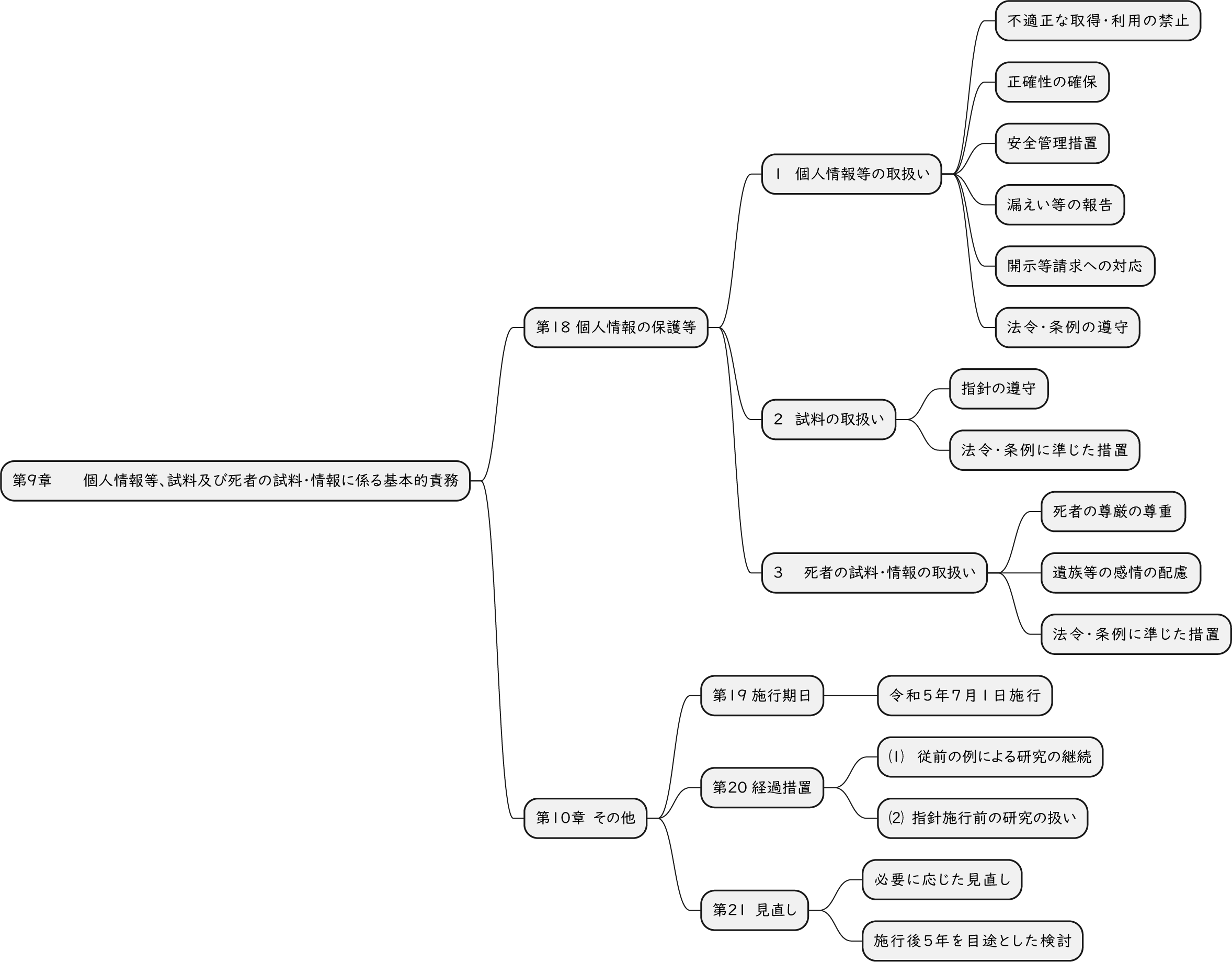
ざっくり何について書かれているのかを把握しやすいようなマインドマップにまとめてみました。情報の抽出にはCopilotを使用しました。ところどころおかしなところがあるのは把握しています。気が向いたら修正したいとは思っていますが、とりあえず、そのまま掲載しています。どこの項に知りたいことが書かれているのかを探す向きにはこういうまとめも有用だろうと思います。実務に重要なポイントは元の文章を確認する必要があります。
もとになったのは令和6年3月27日一部改訂版
pROCパッケージのaSAHデータを使用して、多変量ロジスティック解析を行い、outcomeを他の変数で予測する予測式を作成してみましょう。
以下は具体的な手順です:
まず、必要なパッケージをインストールして読み込みます。
install.packages("pROC")
install.packages("dplyr")
library(pROC)
library(dplyr)
library(ggplot2)
次に、aSAHデータを読み込みます。予測式を作成する際にはカテゴリーを1, 0 の数字に置き換えたほうがいいので、outcome, gender wfnsをダミー変数に変換します。wfinsは各値を別々のダミー変数wfns2, wfns3, wfns4, wfns5に置き換えました。これらすべてが0ならwfns1が1になるはず(欠測値が無い)という事で、wfns1は作成しませんでした。
data(aSAH, package = "pROC")
# outcome変数を数値型のダミー変数に変換
aSAH <- aSAH %>%
mutate(good_outcome = ifelse(outcome == "Good", 1, 0))
# gender変数を数値型のダミー変数に変換
aSAH <- aSAH %>%
mutate(gender_numeric = ifelse(gender == "Male", 1, 0))
# wfns変数のダミー変数を作成し、データフレームに追加
aSAH <- aSAH %>%
mutate(wfns2 = ifelse(wfns == 2, 1, 0),
wfns3 = ifelse(wfns == 3, 1, 0),
wfns4 = ifelse(wfns == 4, 1, 0),
wfns5 = ifelse(wfns == 5, 1, 0))
多変量ロジスティック回帰モデルを作成します。この例では、outcomeを他の変数で予測するモデルを構築します。
# ロジスティック回帰モデルの構築
model <- glm(good_outcome ~ gender_numeric + age + wfns2 + wfns3 + wfns4 + wfns5 + s100b + ndka,
data = aSAH, family = binomial)
# モデルの概要を表示 係数coefを抽出
summary(model)
summary_model <- summary(model)
coef_model <- summary_model$coefficients
# モデル
logit_P = coef_model[1] + coef_model[2]*aSAH$gender_numeric + coef_model[3]*aSAH$age +
coef_model[4]*aSAH$wfns2 + coef_model[5]*aSAH$wfns3 + coef_model[6]*aSAH$wfns4 +
coef_model[7]*aSAH$wfns5 + coef_model[8]*aSAH$s100b + coef_model[9]*aSAH$ndka
aSAH$logit_P <- logit_P
ステップ3までで、モデルに基づいて予測式を作成しました。
# 散布図の作成 データの外観を確認する
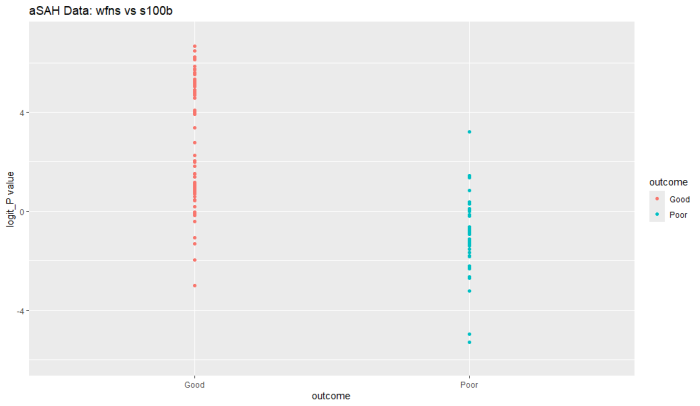
ggplot(aSAH, aes(x=outcome, y=logit_P, color=outcome)) +
geom_point() +
labs(title="aSAH Data: wfns vs s100b",
x="outcome",
y="logit_P value") +
scale_y_continuous(limits = c(-6, 7))
# とりあえずROCカーブを描いてみよう
# roc1 <- aSAH %>% roc(outcome, logit_P)
# ggroc(roc1)
# 散布図の作成 データの外観を確認する
ggplot(aSAH, aes(x=outcome, y=logit_P, color=outcome)) +
geom_point() +
labs(title="aSAH Data: outcome vs logit_P",
x="outcome",
y="logit_P value")+
scale_y_continuous(limits = c(-6, 7))
# ROC曲線の作成 s100b血清値でSAHの予後予測する設定
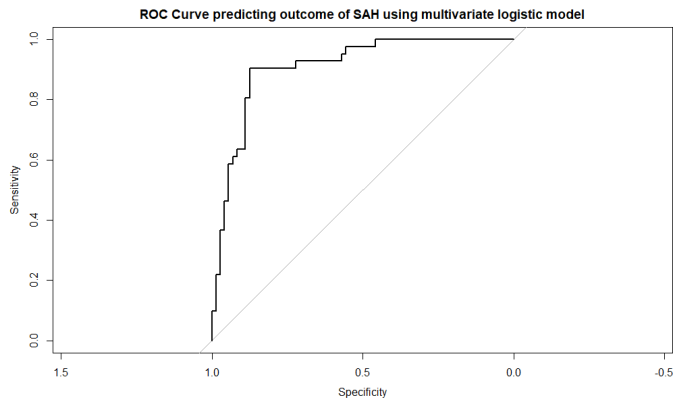
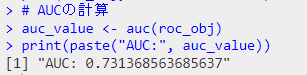
roc_obj <- roc(aSAH$outcome, aSAH$logit_P)
# ROC曲線のプロット
plot(roc_obj, main="ROC Curve predicting outcome of SAH using multivariate logistic model")
# AUCの計算
auc_value <- auc(roc_obj)
print(paste("AUC:", auc_value))
# 感度、特異度、陽性的中率、陰性的中率を計算
coords_obj <- coords(roc_obj, x="best", ret=c("sensitivity", "specificity", "ppv", "npv"))
# 結果を出力
print(paste("感度:", coords_obj["sensitivity"]))
print(paste("特異度:", coords_obj["specificity"]))
print(paste("陽性的中率:", coords_obj["ppv"]))
print(paste("陰性的中率:", coords_obj["npv"]))
# 尤度比を算出、その前に改めて感度と特異度を取得
coords_obj <- coords(roc_obj, x="best", ret=c("sensitivity", "specificity"))
sensitivity <- coords_obj["sensitivity"]
specificity <- coords_obj["specificity"]
# 陽性尤度比 (LR+) と 陰性尤度比 (LR-) を計算
positive_likelihood_ratio <- sensitivity / (1 - specificity)
negative_likelihood_ratio <- (1 - sensitivity) / specificity
# 結果を出力
print(paste("陽性尤度比 (LR+):", positive_likelihood_ratio))
print(paste("陰性尤度比 (LR-):", negative_likelihood_ratio))
# 最適な閾値を取得
best_coords <- coords(roc_obj, x = "best", ret = c("threshold", "sensitivity", "specificity"))
# 最適な閾値を表示
best_coords
データの散布図、教科書のロジスティック回帰のlogitの説明で見たことあるような雰囲気
結果のROC曲線です。結構いい。
その他性能や閾値です

まず、pROCパッケージを使用してROC曲線を作成し、AUCを計算する例を示します。
# パッケージのインストールと読み込み
install.packages("pROC")
library(pROC)
# サンプルデータの読み込み
data(aSAH)
# 散布図の作成 データの外観を確認する
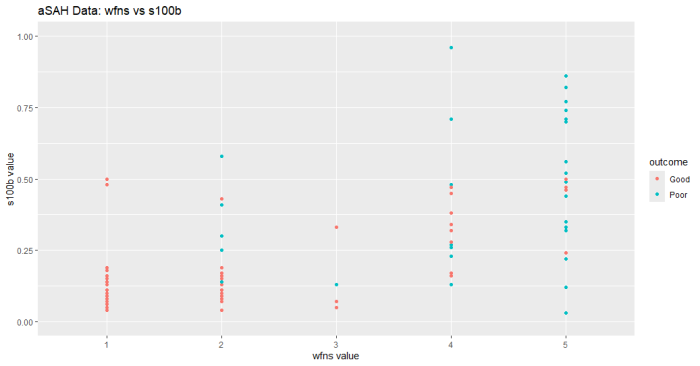
ggplot(aSAH, aes(x=wfns, y=s100b, color=outcome)) +
geom_point() +
labs(title="aSAH Data: wfns vs s100b",
x="wfns value",
y="s100b value")+
scale_y_continuous(limits = c(0, 1))
# ROC曲線の作成 s100b血清値でSAHの予後予測する設定
roc_obj <- roc(aSAH$outcome, aSAH$s100b)
# ROC曲線のプロット
plot(roc_obj, main="ROC Curve predicting outcome of SAH using s100b value")
# AUCの計算
auc_value <- auc(roc_obj)
print(paste("AUC:", auc_value))
# 感度、特異度、陽性的中率、陰性的中率を計算
coords_obj <- coords(roc_obj, x="best", ret=c("sensitivity", "specificity", "ppv", "npv"))
# 結果を出力
print(paste("感度:", coords_obj["sensitivity"]))
print(paste("特異度:", coords_obj["specificity"]))
print(paste("陽性的中率:", coords_obj["ppv"]))
print(paste("陰性的中率:", coords_obj["npv"]))
# 尤度比を算出、その前に改めて感度と特異度を取得
coords_obj <- coords(roc_obj, x="best", ret=c("sensitivity", "specificity"))
sensitivity <- coords_obj["sensitivity"]
specificity <- coords_obj["specificity"]
# 陽性尤度比 (LR+) と 陰性尤度比 (LR-) を計算
positive_likelihood_ratio <- sensitivity / (1 - specificity)
negative_likelihood_ratio <- (1 - sensitivity) / specificity
# 結果を出力
print(paste("陽性尤度比 (LR+):", positive_likelihood_ratio))
print(paste("陰性尤度比 (LR-):", negative_likelihood_ratio))

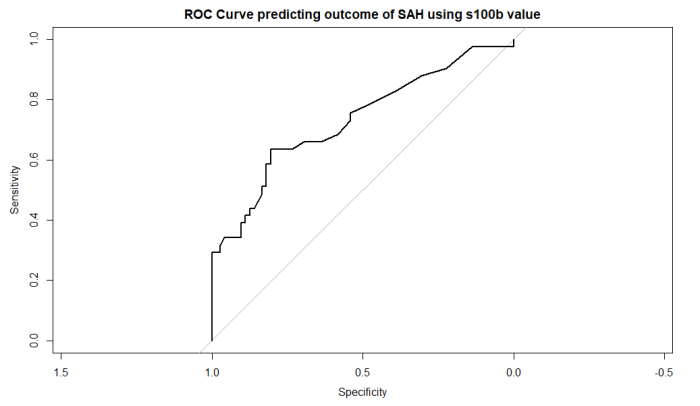
s100bでoutcomeを予測する予後予測の性能としての計算


このパッケージ、パイプを使うと、だいぶシンプルで見やすくなります
ここでは予測に上述のs100bの代わりにwfnsを使用してみました
data(aSAH)
library(dplyr) #パイプを使用するためにパッケージを使用
roc2.1 <- aSAH %>% roc(outcome, wfns)
ggroc(roc2.1)
| 関数名 | 説明 | 使用例 |
|---|---|---|
roc() | ROC曲線を作成 | roc(response, predictor) |
plot() | ROC曲線を描画 | plot(roc_object) |
auc() | ROC曲線のAUCを計算 | auc(roc_object) |
ci() | ROC曲線の信頼区間を計算 | ci(roc_object) |
coords() | ROC曲線上の特定の点の座標を取得 | coords(roc_object, "best") |
ggroc() | ggplot2を使用してROC曲線を描画 | ggroc(roc_object) |
roc.test() | ROC曲線の統計的な比較 | roc.test(roc_object1, roc_object2) |
smooth() | ROC曲線を平滑化 | smooth(roc_object) |
var() | ROC曲線のAUCの分散を計算 | var(roc_object) |
次に、ROCRパッケージを使用してROC曲線を作成し、パフォーマンスを評価する例を示します。
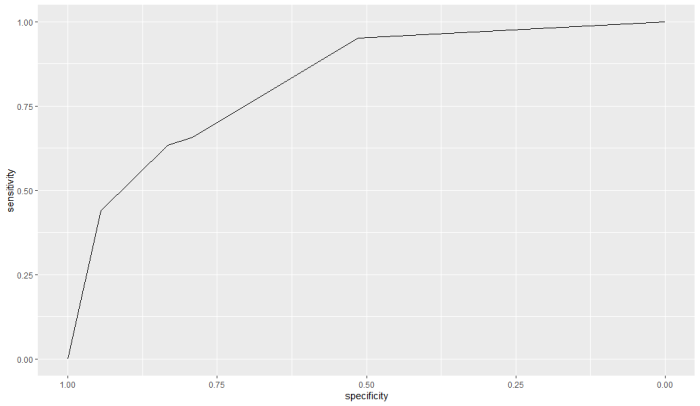
その前にiris データをプロット。ここではPetalLengthが5以上でvirginicaと判断する予測方法のパフォーマンスを調べるようにします
# 基本パッケージの読み込み
library(ggplot2)
# irisデータセットの読み込み
data(iris)
# 散布図の作成
ggplot(iris, aes(x=Petal.Length, y=Petal.Width, color=Species)) +
geom_point() +
labs(title="Iris Data: Petal Length vs Petal Width",
x="Petal Length",
y="Petal Width")
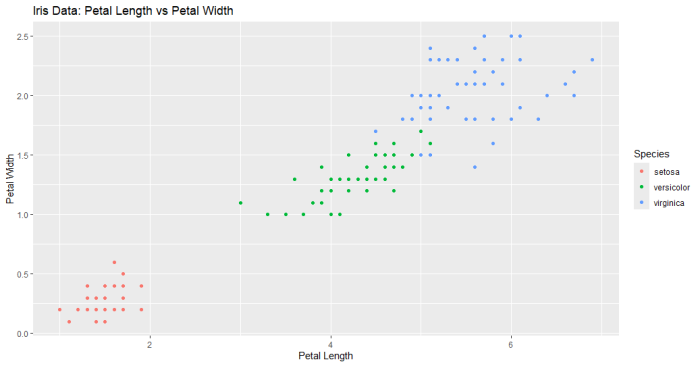
# パッケージのインストールと読み込み
# install.packages("ROCR")
library(ROCR)
# irisデータセットの読み込み
data(iris)
# 二値分類のためのデータ準備
iris_binary <- iris
iris_binary$Species <- ifelse(iris$Species == "virginica", 1, 0)
# 予測値としてPetal.Lengthを使用し、閾値5で二値化
predictions <- ifelse(iris_binary$Petal.Length >= 5, TRUE, FALSE)
labels <- iris_binary$Species
# Predictionオブジェクトの作成
pred <- prediction(as.numeric(predictions), labels)
# パフォーマンスオブジェクトの作成
perf <- performance(pred, measure = "tpr", x.measure = "fpr")
# ROC曲線のプロット
plot(perf, main="ROC Curve with Petal Width Threshold")
# AUCの計算
auc_value <- performance(pred, measure = "auc")
auc_value <- auc_value@y.values[[1]]
print(paste("AUC:", auc_value))
結果のプロット
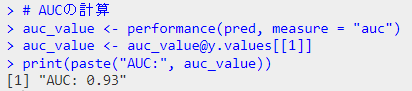
結果が奇麗すぎ(データのプロット見たらわかるように、一つの変数で種をきれいに分離できますからね)

表題のキーボードの設定を簡単に解説
・ 設定用アプリ「MINI KeyBoard.exe」を起動

・ 「危険」と言う様な警告画面が出ますが、心配しつつ実行
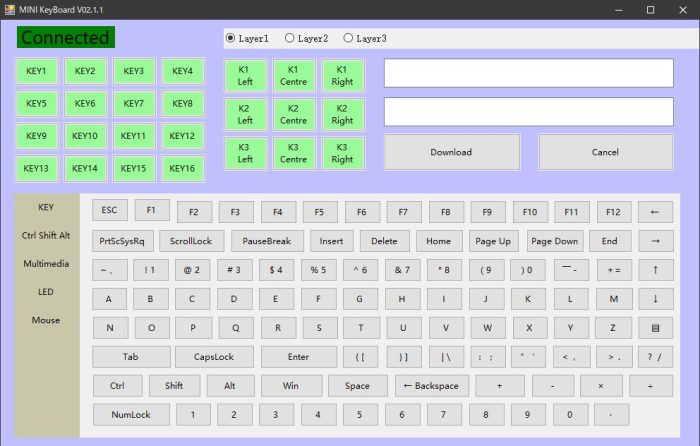
・ Connectedの下のKEY1をタップして、ハイライトして、設定したいキーを選ぶ(図ではBackspaceを選択)
・ Download (キーボードへ設定する情報を送信する意味の様です図では赤まる)をタップ
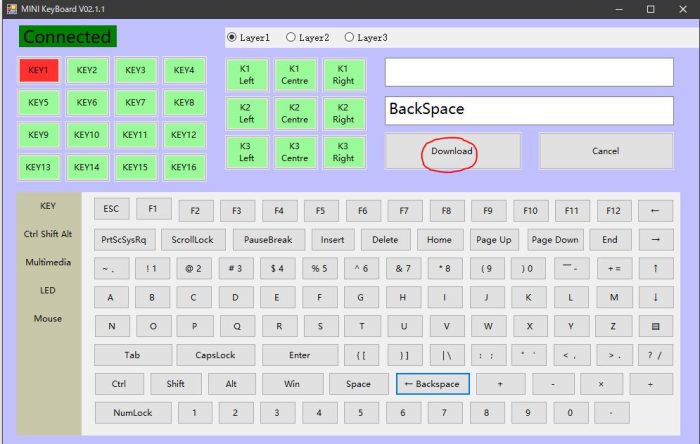
同様にKEY2, KEY3も設定する。
—
以下はプログラマブルキーボード(3キー)のマニュアルの機械翻訳です
このリンク先にオリジナルの情報とアプリが保存されています。一応ダウンロードして、ウイルススキャンを行って、2025/1/29 時点のファイルには問題のある罠は仕掛けられていない事を確認しています。将来、誰かが何かを仕掛けることもあり得ますので、ダウンロードしたら毎回チェックは必要でしょう。
Romoral ユーザーマニュアル —3キー有線RGBバージョン用
注意: このソフトウェアはWindowsシステムでのみ動作します。PC側のコンピュータでダウンロードしてください。設定ソフトウェアはインストール不要で、開いた後に直接実行できます。設定後はキーボードに自動的に保存され、LinuxやMacOSなどのUSBプロトコルをサポートするコンピュータに接続して使用できます。(初回はウイルス対策ソフトに注意し、信頼できるファイルとして設定してください)!!
上記の手順に従って進めてください。同時に、ソフトウェアパッケージにはユーザーマニュアル、ショートカットキー設定ビデオ、日常の質問への回答が含まれています。インストール中に質問が発生した場合は、心配せずに問題のスクリーンショットやビデオを送ってください。サポートいたします。ありがとうございます。
フレンドリーフィードバックを残してくださった購入者様へ: 生涯無料の相談とサービスを提供します。また、オリジナルのシャフトスイッチをギフトとして提供します。(新しい注文と一緒に発送できます)。このギフトは生涯有効です。請求されるまで有効です。ありがとうございます。(Choiceストアの購入者様は、Romoralの公式ストアカスタマーサービスに連絡して受け取ってください)
製品について
日常の質問への回答

前回の記事では、一般的な従来使用されてきたという意味でのワクチンのシェディングの例を文献を引用して説明しました。そして、レプリコンワクチンに対する懸念として拡散されている「シェディング」が従来使用されていたのとは違う意味だという点、根拠とされるSeneff氏の文献は直接の根拠を示さず、引用文献を引いているのみだという事を記載しました。今回の記事ではSeneff氏が根拠として引用しているLucchetti氏の論文(Lucchetti et al., 2021)を見てゆきます。
Detection and characterisation of extracellular vesicles in exhaled breath condensate and sputum of COPD and severe asthma patients
直訳すると「COPDおよび重症喘息患者の呼気凝縮液および痰中の細胞外小胞の検出と特性評価」
タイトルを見ますと、mRNAワクチンの何かを論じているものとは読み取れません。タイトルだけみても慢性閉塞性肺疾患あるいは重症ぜんそく患者の呼気や喀痰に細胞外小胞が検出できたという話だとわかります。
Lucchetti氏の論文(Lucchetti et al., 2021)の掲載されていますEuropian Respiratory Journalは直近のインパクトファクターが16.6と一流雑誌という事ができます。
学術雑誌の記事には、論文が専門家によって査読されて後に世に出るピアレビュー誌とそうでない非ピアレビュー誌があります。「ピアレビュー誌」の中にも専門家によって査読される記事とそうでない記事があります。詳しくは各雑誌の投稿規定を見ると書かれていますが、「オリジナルアーティクル」は通常ピアレビューの記事になります。一方、「エディトリアル」と呼ばれる、当該雑誌に掲載されたオリジナルアーティクルの紹介文や編集者からのメッセージ、それに、訂正のお知らせなどはピアレビューではありません。専門家同士のコミュニケーションである、レターやショートコミュニケーションはピアレビューを実施する雑誌とそうでない雑誌がありますが、ピアレビューを実施しない雑誌でも編集者が目を通して一定の基準を満たしたものが掲載されることが多いはずです。
このLucchetti氏の論文はResearch Letterの記事で、記事のボリュームやデータがしっかり示されて論述しているところからはピアレビューを受けて世に出されたものだと思われます。
以下が当該論文の概要です:
上記の通りLucchetti氏の論文は慢性閉塞性肺疾患(COPD)か重症喘息患者を対象としていて、mRNAワクチンの接種の有無は問題にしていません。直径20-1000nmほどの小型の膜で覆われた小胞の事を問題にしています。これはコロナウイルスのサイズ100nm付近に近いレンジで、スパイクタンパク(10nmほどか?)は議論に出てきていません。少なくとも直接的にmRNAワクチンを接種したヒトからスパイクタンパクが放出されるという事を示したという話ではありません。
私の読み解いたところでは、Lucchetti氏の論文は普通の学術論文で、一流紙で論じられた科学的な知見であるが、それはmRNAワクチンのシェディングを懸念するというデータではありません。この部論文を引用したSeneff氏が持論に都合良いように曲解しているとなります。
Lucchetti, D., Santini, G., Perelli, L., Ricciardi-Tenore, C., Colella, F., Mores, N., Macis, G., Bush, A., Sgambato, A., & Montuschi, P. (2021). Detection and characterisation of extracellular vesicles in exhaled breath condensate and sputum of COPD and severe asthma patients. 58(2), 2003024. https://doi.org/10.1183/13993003.03024-2020

COVID-19およびそのワクチンに関する情報を目にする中で、比較的知識があると思われる方々でさえ、いわゆる陰謀論や科学的根拠の乏しい情報に惑わされているように見受けられます。これは一つには、疫学データの解釈が非常に難しいことに起因しています。一見簡単に見えるデータであっても、疫学特有の手法や癖により誤解を生むことがあります。異業種で経験を積んだ方々が疫学データを解釈する際、疫学特有の罠に陥り、独自の理論を展開してしまうことがしばしば見受けられます。
したがって、情報源としては、疫学研究の実績があり、疫学データの解釈に豊富な経験を持つ専門家、あるいは実際の臨床現場で当該診療領域の経験を持つ専門家の発言を重視する必要があります。用語一つをとっても、その領域での使用方法には歴史があり、特定の意味があります。用語だけを流用し、具体的な意味が曖昧なまま使用することは避けるべきです。例えば、インターネット上で見かける「シェディング」という言葉の使い方についても、注意深く検討する必要があります。
ワクチン接種が進む中で、「ワクチンのシェディング」という言葉を耳にすることが増えてきました。これは一体何を意味するのでしょうか?一般の方々にとっては少し難しい概念かもしれませんが、ここではできるだけ分かりやすく説明してみたいと思います。
一般的にはワクチンのシェディングとは、ワクチンを接種した人が、ワクチン由来のウイルス粒子を体外に排出する現象を指します。これは特に生ワクチンに関連して見られることが多いです。例えば、ロタウイルスワクチンに関する研究では、シェディングが一般的であり、長期間にわたって続くことがあると報告されています。
具体的な例を挙げると、五価ロタウイルスワクチン(RV5)の初回接種後、21.4%の乳児で最大9日間にわたってシェディングが確認されました(Yen et al., 2011)。また、別の研究では、初回接種後5〜10日で93%の乳児がワクチン関連の粒子を排出し、その中でG1という遺伝子型が主に見られました (Markkula et al., 2019)。
RV5とRotarix(RV2)という2種類のロタウイルスワクチンを比較した研究では、シェディングの割合は似ているものの、RV2を接種した人の方がより多くのウイルスを排出していることが分かりました(Hsieh et al., 2014)。
一方で、経口狂犬病ワクチン(SPBN GASGAS)に関する研究では、様々な動物種において24時間以上感染性ウイルスの活発なシェディングは見られず、水平感染のリスクは最小限であることが示唆されています (Vos et al., 2018)。
これらの研究結果は、ワクチンの種類や接種を受ける種によって、シェディングのパターンが大きく異なることを示しています。ワクチンのシェディングについて理解を深めることで、ワクチン接種に関する不安を軽減し、より安心して接種を受けることができるでしょう。
世の中で数多くの医学・生物学の論文が公開されているのですが、その多くの論文を網羅的に検索できるPubMedと言う米国NLMが提供するデータベースがあります。多くの研究者がPubMedを利用しており、また、論文を投稿する際には自信の研究を世に広く認めてもらおうと、PubMedに登録される雑誌に論文を掲載するように投稿先を考えます。私がそのPubMedを利用して調査した限りでは、新型コロナウイルスワクチンに関するシェディングが発生したことを示す学術論文は見つかりませんでした。生ワクチンとは異なり、現在日本で広く接種されているmRNAワクチンや遺伝子組換えタンパク質ワクチンは、ウイルスの一部の成分のみを使用しているため、ワクチン由来の遺伝子だけではウイルス粒子を構築するために必要な構成要素が揃いません。このため、ワクチン由来のウイルス粒子が構築されることは理論的に考えにくいです。
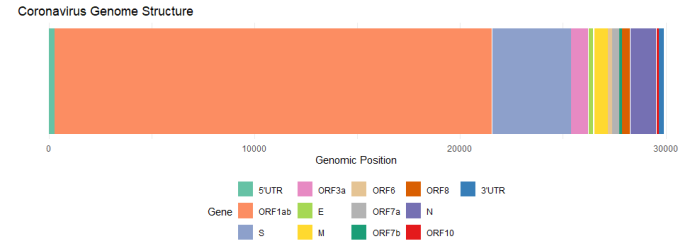
下の図は報告されたSARS-CoV-2ウイルス起源株の遺伝子配列(Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, co – Nucleotide – NCBI (nih.gov))を基に遺伝子の配置を色分けしたものです。図ではORF1abを一つの色で塗っていますが、ORF1abの領域にはさらにいくつかのタンパク質をコードする領域が含まれています。多くの遺伝子が揃って初めてウイルス粒子としての構造が構成されるため、Sタンパク質をコードする遺伝子だけでウイルス粒子の構造を形成することは難しいと考えられます。
それにもかかわらず、レプリコンワクチンにシェディングの懸念があると主張するウェブサイトが存在しますが、根拠データは直接は示されていません。当該声明では、Seneff, S., & Nigh, G. (2021). Worse than the disease? Reviewing some possible unintended consequences of the mRNA vaccines against COVID-19. International Journal of Vaccine Theory, Practice, and Research, 2(1), 38-79.の文献を引用していますが、この文献はPubMedには掲載されていないため、検索にかかりません。
Seneff氏の所属はComputer Science and Artificial Intelligence Laboratory, MIT, Cambridge MA, 02139, USAであり、ウイルスやワクチンの専門施設ではありません。文献内にも、実験や臨床試験で直接ウイルスのシェディングを確認したデータは記載されていません。また、ワクチンの領域で問題となっているのはウイルス粒子のシェディングですが、Seneff氏の文献ではタンパク質のシェディングについて論じています。膜タンパク質が細胞膜表面で切断されて細胞から離れる現象をシェディングと呼ぶ研究領域もありますが、人から人への伝播を問題にしているため、膜タンパク質のシェディングとは異なる意味合いです。根拠の有無は引用文献を確認する必要がありますが、仮にスパイクタンパク質が被接種者から放出されたとしても、それがウイルスとして感染し、COVID-19を発病するとは考えにくいです。
Seneff氏は、従来のワクチンの安全性で問題になるシェディングとは異なる意味で「シェディング」を使用していることが明らかになりました。異業種間で同じ言葉を異なる意味で使用することはあり得るため、その言葉の共通認識を持たない限り議論は成立しません。まず、この研究者がどのようなデータを基に、どのような現象を「シェディング」と称しているのかを明確にする必要があります。
もう一点気になる点があります。それはSeneff氏の論文はmRNAワクチンを対象にして記述されている点です。レプリコンワクチンでの懸念に直に結びつけて良いものか。少なくとも、従来のRNAワクチンでは問題としなかったにもかかわらず、レプリコンワクチンでは懸念であるとするのであれば、違うワクチンについての情報を引用して根拠とすることの正当性の説明は見てみたいところです。
Seneff氏は、レプリコンワクチンについて論じている訳ではないのですが、mRNAワクチンでの「スパイクタンパク質のシェディング」を論じていました。その直接的な根拠となるような観察の記録データは記載されておらず、代わりにLucchetti氏らの論文(Lucchetti et al., 2021) を引用しています。Lucchetti氏らの論文はPubMedに掲載されていますので、この記事でもReferencesの中に書誌事項を記載しておきます。Lucchetti氏らの論文ではどのような研究がなされているのかは別記事にしようと思いますが、このSeneff氏の文献のシェディング周りの部分をもう少しだけ見ておこうと思います。

この部分を機械翻訳しました。
インターネット上では、ワクチン接種者が近くにいる未接種者に病気を引き起こす可能性についての議論が多くあります。これは信じがたいかもしれませんが、脾臓の樹状細胞から誤って折りたたまれたスパイクタンパク質を含むエクソソームが放出され、他のプリオン再構成タンパク質と複合体を形成することで起こり得るというもっともらしいプロセスがあります。これらのエクソソームは遠くまで移動することができます。肺から放出され、近くの人が吸い込むことも不可能ではありません。エクソソームを含む細胞外小胞は、痰、粘液、上皮内液、気管支肺胞洗浄液において呼吸器疾患と関連して検出されています(Lucchetti et al., 2021)。
「もっともらしいプロセス」があると言い切っています。それぞれの現象があるとして、そのプロセスが実際に起こるのか、想像の中でつないだだけなのかはわからないものなのですが、新型コロナウイルスのワクチンを接種したヒトに実際に起きたというデータは明記されていません。あと、プリオンと言えばクロイツフェルト・ヤコブ病や狂牛病を引き起こすことで知られていますが、mRNAワクチンの副反応や新型コロナウイルスの病態に関連しているという話は聞いたことがありません。
私がここまで見てきた範囲では、Seneff氏の文献は、自然現象や実験環境での現象を観察して、その観察結果を解釈するという科学的なプロセスに基づいたロジックは読み取れない文書であることが理解できました。
References:
Hsieh, Y.-C., Wu, F.-T., Hsiung, C. A., Wu, H.-S., Chang, K.-Y., & Huang, Y.-C. (2014). Comparison of virus shedding after lived attenuated and pentavalent reassortant rotavirus vaccine. 32(10), 1199–1204. https://doi.org/10.1016/j.vaccine.2013.08.041
Lucchetti, D., Santini, G., Perelli, L., Ricciardi-Tenore, C., Colella, F., Mores, N., Macis, G., Bush, A., Sgambato, A., & Montuschi, P. (2021). Detection and characterisation of extracellular vesicles in exhaled breath condensate and sputum of COPD and severe asthma patients. 58(2), 2003024. https://doi.org/10.1183/13993003.03024-2020
Markkula, J., Hemming-Harlo, M., & Vesikari, T. (2020). Shedding of oral pentavalent bovine-human reassortant rotavirus vaccine indicates high uptake rate of vaccine and prominence of G-type G1. 38(6), 1378–1383. https://doi.org/10.1016/j.vaccine.2019.12.007
Vos, A., Freuling, C., Ortmann, S., Kretzschmar, A., Mayer, D., Schliephake, A., & Müller, T. (2018). An assessment of shedding with the oral rabies virus vaccine strain SPBN GASGAS in target and non-target species. 36(6), 811–817. https://doi.org/10.1016/j.vaccine.2017.12.076
Yen, C., Jakob, K., Esona, M. D., Peckham, X., Rausch, J., Hull, J. J., Whittier, S., Gentsch, J. R., & LaRussa, P. (2011). Detection of fecal shedding of rotavirus vaccine in infants following their first dose of pentavalent rotavirus vaccine. 29(24), 4151–4155. https://doi.org/10.1016/j.vaccine.2011.03.074

Vaccine refusal, particularly for COVID-19, is a complex issue influenced by various factors. Studies suggest that refusal may be reinforced by informational rewards, increased self-esteem, and tribal identity (Brakel & Foxall, 2022). Some argue that vaccine refusers can be epistemically rational and responsible for their beliefs, challenging the legitimacy of compulsory vaccination policies (Meylan & Schmidt, 2023). Research in Ethiopia found that factors such as age, perception of the vaccine, eHealth literacy, information sources, and internet use significantly influence vaccine acceptance (Kalayou & Awol, 2022). Myths, conspiracy theories, and misinformation spread through various channels contribute to vaccine hesitancy, potentially impacting global vaccine programs beyond COVID-19 (Ullah et al., 2021). These findings highlight the multifaceted nature of vaccine refusal and the need for targeted interventions to address hesitancy, considering factors such as information sources, digital literacy, and individual perceptions.
結論とか議論の意味は分かるものの、この領域の研究手法は私の理解が及ばないところ
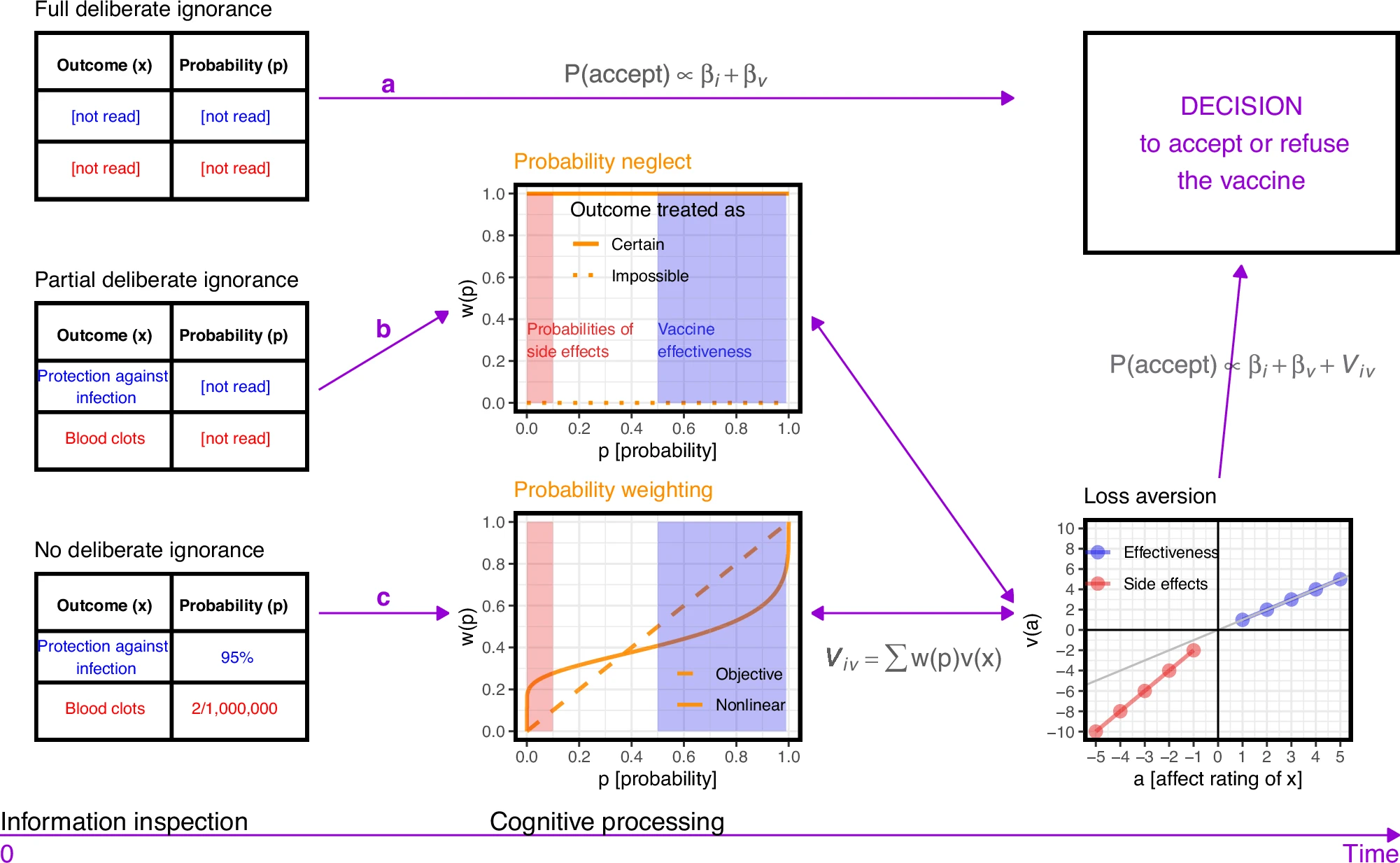
ワクチンの証拠に関する情報を処理するさまざまな方法により、個々の i がワクチン v を受け入れる確率 (P(accept) で表されます)。パスaは、完全な意図的な無知を表しています。ワクチンの証拠はまったく検査されず、決定は、個々のiとワクチンv(βで示される)に関連する他の要因に基づいています
βiv.
パスbは確率無視を表しており、ワクチンの可能な結果のみが獲得され、その確率は獲得されない部分的な意図的な無知の一種です。このような場合、人々は通常、結果が確実に起こると認識します。しかし、原理的には、確率値を無視して、対応する結果が発生することは不可能であると認識することも可能である。パスcは意図的な無知を表していません。すべての情報が検査されますが、確率情報は非線形確率重み付けによって認知的に歪められる可能性があります。確率重み付け関数の曲率は、このような歪みの程度を測定します。パス (b) から (c) では、無視された確率と重み付けされた確率 w(p) は、対応する結果の主観的値と統合され、モデルでは影響評価 a によって数値的に表され、値関数 v で変換されます。ワクチンの副作用とベネフィットに対する価値関数の傾きの差は、損失回避の尺度を構成し、これは私たちの調査で考慮された2番目の認知の歪みです。
まとめ:
ワクチン拒否の問題を解決するためには、情報源やデジタルリテラシー、個々の認識などを考慮した対策が必要です。
Vaccine Refusal: A Preliminary Interdisciplinary Investigation – PubMed (nih.gov)
Full article: Refusing the COVID-19 vaccine: What’s wrong with that? (tandfonline.com)

What are the molecular targeted drugs for cancers with BRCA1 and BRCA2 gene mutations?
BRCA1およびBRCA2遺伝子変異を持つがんに対する分子標的薬で注目されているのはPARP阻害剤(PARPi)です。現在、FDAに承認されているPARPiは4種類あり、これらはBRCA1/2欠損がんの治療に利用されています(Ragupathi et al., 2023)。
最初に承認されたPARPiであるオラパリブは、BRCA変異を持つ卵巣がん患者の治療において非常に有望な結果を示しています(Venkitaraman, 2009; Tangutoori et al., 2015)。PARPiは、がん細胞のゲノム不安定性を利用してDNA損傷応答を標的とし、従来の化学療法と比較してより腫瘍細胞選択的なアプローチを提供します(O’Connor, 2015)。
PARPiの細胞毒性は、BRCA1/2変異腫瘍の複製が困難なゲノム領域に過剰な複製ストレスを誘導することによると考えられています(Ragupathi et al., 2023)。現在進行中の研究では、PARPiと免疫チェックポイント阻害薬を組み合わせて臨床結果を向上させる方法が探求されています(Ragupathi et al., 2023)。さらに、PARPiは他のさまざまながんタイプに対しても、単独療法および他の治療法との併用療法としての利用が検討されています(Tangutoori et al., 2015)。
このように、PARP阻害剤はBRCA1/2遺伝子変異を持つがん患者にとって新たな希望となる治療法であり、今後の研究と臨床応用が期待されています。
Molecular targeted drugs for cancers with BRCA1 and BRCA2 gene mutations primarily focus on PARP inhibitors (PARPi). Four FDA-approved PARPi are currently available for treating BRCA1/2-deficient cancers (Ragupathi et al., 2023). Olaparib, the first approved PARPi, has shown promise in treating ovarian cancer patients with BRCA mutations (Venkitaraman, 2009; Tangutoori et al., 2015). PARPi exploit the genomic instability of cancer cells by targeting the DNA damage response, offering a more selective approach compared to traditional chemotherapy (O’Connor, 2015). The cytotoxic effect of PARPi is believed to result from inducing excessive replication stress in difficult-to-replicate genomic regions of BRCA1/2 mutated tumors (Ragupathi et al., 2023). Ongoing research explores combining PARPi with immuno-oncology drugs to enhance clinical outcomes (Ragupathi et al., 2023). Additionally, PARPi are being investigated for use in various other cancer types, both as monotherapies and in combination with other treatments (Tangutoori et al., 2015).
References
O’Connor, M. J. (2015). Targeting the DNA Damage Response in Cancer. 60(4), 547–560. https://doi.org/10.1016/j.molcel.2015.10.040
Ragupathi, A., Singh, M., Perez, A. M., & Zhang, D. (2023). Targeting the BRCA1/2 deficient cancer with PARP inhibitors: Clinical outcomes and mechanistic insights. 11, 1133472. https://doi.org/10.3389/fcell.2023.1133472
Tangutoori, S., Baldwin, P., & Sridhar, S. (2015). PARP inhibitors: A new era of targeted therapy. 81(1), 5–9. https://doi.org/10.1016/j.maturitas.2015.01.015
Venkitaraman, A. R. (2009). Targeting the molecular defect in BRCA-deficient tumors for cancer therapy. 16(2), 89–90. https://doi.org/10.1016/j.ccr.2009.07.011
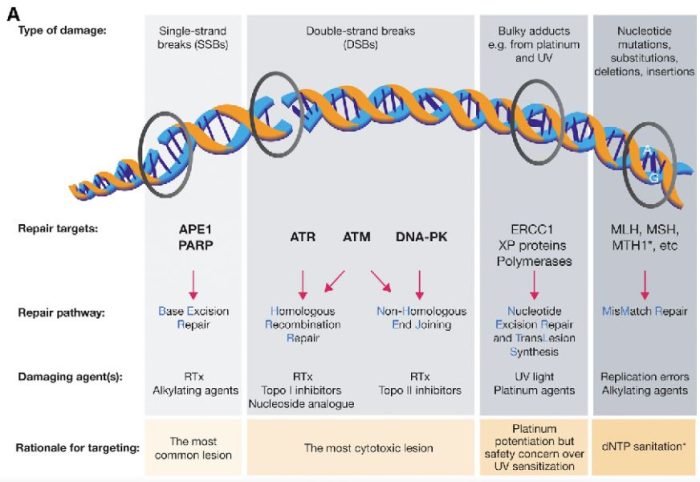
図の解説:PARP(ポリ(ADP-リボース)ポリメラーゼ)は、主に一重鎖DNA切断(single-strand breaks, SSB)の修復に関与しています。具体的には、以下のようなDNAダメージを修復します:
PARPは、これらの損傷を修復することで、細胞のゲノム安定性を維持し、細胞の生存を助けます。しかし、BRCA1やBRCA2遺伝子に変異がある場合、これらの修復経路が正常に機能しないため、PARP阻害剤(PARPi)はこれらのがん細胞に対して特に効果的です。PARPiはPARPの機能を阻害することで、がん細胞に蓄積するDNA損傷を増加させ、最終的にがん細胞の死を誘導します。
PARP分子と、Olaparibの結合を結晶構造で眺めてみました。この構造だと、PARPたんぱく質に結合したOlaparibは分子の中に深く埋もれているように見えます。(結合した後にたんぱく質の構造変化が起きる?)緑色の分子がolaparibで、それ以外が human PARPのcatalytic domain。下図は下記文献のデータを基にわたくしが作図しました。
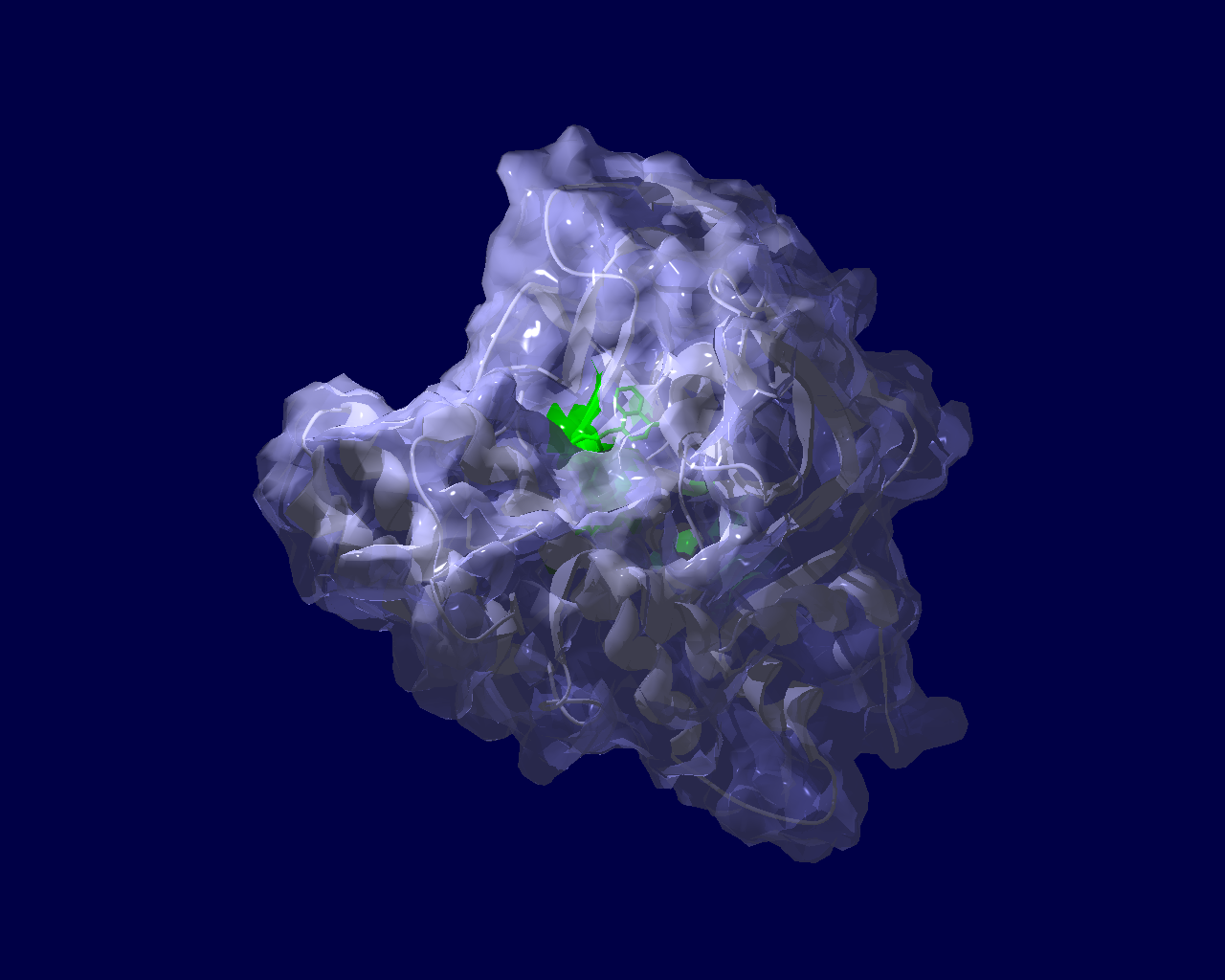
Ogden, T. E. H., Yang, J.-C., Schimpl, M., Easton, L. E., Underwood, E., Rawlins, P. B., McCauley, M. M., Langelier, M.-F., Pascal, J. M., Embrey, K. J., & Neuhaus, D. (2021). Dynamics of the HD regulatory subdomain of PARP-1; substrate access and allostery in PARP activation and inhibition. 49(4), 2266–2288.
https://doi.org/10.1093/nar/gkab020

Mpox(旧称:monkeypox)は、天然痘に似た症状を呈するが、より軽度なウイルス性人獣共通感染症です(Jarrell & Perryman, 2023)。動物から人間へ、また人間同士の直接接触、呼吸器飛沫、汚染物を介して伝播することがあります(Jarrell & Perryman, 2023)。2022年の最近の世界的な流行により、WHOはこれを国際的に懸念される公衆衛生上の緊急事態と宣言しました(Afzal, 2023; Aden et al., 2023)。Mpoxは通常、発熱性の発疹性疾患として現れ、ポリメラーゼ連鎖反応検査によって診断が確認されます(Kumar et al., 2023; Aden et al., 2023)。予防および曝露後予防のために、JYNNEOS®およびACAM2000®の2つのワクチンが利用可能です(Jarrell & Perryman, 2023)。ほとんどの症例は自己制限的ですが、テコビリマット、ブリンシドフォビル、およびシドフォビルなどの抗ウイルス治療がリスクのある集団に対して利用可能です(Jarrell & Perryman, 2023; Afzal, 2023)。この流行は、変化した伝播パターンと世界的な準備および対応努力の必要性についての懸念を引き起こしました(Kumar et al., 2023; Aden et al., 2023)。
Aden, D., Zaheer, S., Kumar, R., & Ranga, S. (2023). Monkeypox (Mpox) outbreak during COVID‐19 pandemic—Past and the future. 95(4), e28701. https://doi.org/10.1002/jmv.28701
Afzal, M. F. (2023). MPOX: A RE-EMERGING INFECTION. 34(02), 58–59. https://doi.org/10.51642/ppmj.v34i02.608
Jarrell, L., & Perryman, K. (2023). Mpox (monkeypox): Diagnosis, prevention, and management in adults. 48(4), 13–20. https://doi.org/10.1097/01.NPR.0000000000000025
Kumar, S., Guruparan, D., & Karuppanan, K. (2023). Recent Advances in Monkeypox (Mpox): Characterization, Diagnosis, and Therapeutics – A Multidimensional Review. https://www.semanticscholar.org/paper/Recent-Advances-in-Monkeypox-(Mpox)%3A-Diagnosis%2C-and-Kumar-Guruparan/f14cc79d55402024d5d362dfcdb18c9d9947170b
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}