




遺伝子治療
AAV中和抗体保持者に対するIgG分解酵素薬VTX-PIDによる第1相治験
VTX-PID as a novel recombinant immunoglobulin G–degrading enzyme (IdeS) for efficient AAV-based gene therapy in participants with neutralizing antibodies: results of the phase I first-in-human NAVIgATE study
Benichou et al. Front. Immunol. 17:1824802 (2026)
AAVを用いた遺伝子治療の障壁の一つに中和抗体があります。AAVはヒトに対して基本的に非病原性であり、日常生活の中で暴露されることが多いことから、成人では多くの人が知らず知らずに抗AAV抗体を保持しております。こういった方々はAAVの全身投与による治療をしても、AAVが中和されてしまい、その効果を期待できないことから、通常治療対象から除外されてしまいます。実際にこの研究が行われたドイツでは、600人弱のスクリーニングで、67%が抗体陰性、19%が中等度、14%が高度抗体保持者だったとのことです。
本研究では、この抗AAV抗体保持者に対するAAV遺伝子治療を可能にするための前処置薬の開発に関する論文です。ここで用いられているのは、Streptococcus pyogenes(化膿レンサ球菌)由来の組換えimlifidase(IdeS)という特異的IgG分解酵素です。この薬剤(VTX-PID)はIdefirix®という名称で腎臓移植時の脱感作療法として使用されているものですが(本邦では未承認)、本研究はこの薬剤をAAV遺伝子治療において一時的に中和抗体を除去し、その間にAAV投与を行うことができるかについて検証するための第1相治験です。興味深い点は、抗AAV中和抗体がほとんどIgGであり、IdeSはIgG以外の免疫グロブリンに対して作用しないため、一過性にIgGを分解除去することで他の免疫系に大きな影響を及ぼすことなく、全身投与されたAAVを標的細胞に送り届けることができるというロジックです。今回は安全性のほか、至適用量の決定などが行われましたが、AAV投与自体は行われておりません。対象者は中等度から高度の抗AAV3B血清型抗体を保持する35名の正常成人男性で、VTX-PIDの投与量を段階的に増加させる(0.075~0.6 mg/kg)か、プラセボが投与されました。ここでは詳細な結果の記述は割愛しますが、全ての用量で大きな副作用を生ずることはなく、安全性が確認されました。抗AAV抗体は大幅に低下し、その後徐々に完全に回復し、高用量群で2−5日間の効果持続が確認されました。中等度の抗AAV3B抗体保持者では全員で中和抗体が非阻害性レベルまで低下しましたが、高度の抗AAV3B抗体保持者では低下レベルは十分ではありませんでした。従って本薬は、中等度の抗AAV抗体保持者に対して、最大5日間の治療機会の窓を開く有望な前処置アプローチであることが示され、全身投与によるAAV遺伝子治療の適格性を拡大すると期待されます。本薬剤は、すでに腎臓移植で前処置薬として臨床使用されていることから、AAV治療での実用化の障壁は低いと思われますので、AAV遺伝子治療の制約を一つ取り払うことができる手法として期待されます。
文責:井上 健
DOI: https://doi.org/10.3389/fimmu.2026.1824802
希少疾患に対する細胞・遺伝子治療の支払い手段としての証券化:シミュレーション研究
Securitization as a means to pay for cell and gene therapies for orphan diseases: a simulation study
Lu et al. Gene Therapy (2026)
今回は少し毛色の変わった論文をご紹介します。どちらかというと医療経済学的なお話です。画期的な遺伝子細胞治療法が今後次々に医療現場に登場することが期待されていますが、特に患者数が少ない超希少疾患については、そのコストの高さや長期効果の不確実性など経済的な理由により実用化が進まないことが問題となっています。今回の論文は、この問題を解決する1つの手段を提案しています。通常の慢性疾患治療は、長期に渡る治療の経過にそって費用負担が生じ、その経済的負担は時間的に分散されています。一方、遺伝子治療では、多くは1回のみの治療で完結し、費用の支払いも1回で多額となりますが、治療効果は長期間に継続します。このズレが存在するため、その効果が不十分である場合には、保険者の支払額は過大となり、効果が大きくても製薬企業にはインセンティブがない、という状況が生じています。また米国では、保険会社が生涯で変わるため、治療を受けたときの保険者のみがその費用負担をしなければなりません。現在、製薬企業は5年間の成果連動型分割払い(performance-based annuity: PBA)という手法を提案していますが、期間が短く、長期効果の不確実性に対応できない、という課題を残しています。そこで筆者らは30年間の成果連動年金型支払い(長期PBA)と金融の証券化(securitization)を組み合わせる新しい支払いモデルを提案しています。これは30年間、患者が生存している限り保険者の支払いを継続する、いわば30年ローンです。さらに製薬企業がPBA契約を締結し、証券化することで、将来の分割支払いを投資家に売却することが可能となり、投資家が前払い資金を提供することで、製薬企業は早期に収益回収が可能になるというものです。これを複数の患者についてプール化し、治療効果を裏付けとした証券Cure-backed securities(CBS)として金融商品するというものです。その構造として、シニア債(低リスク)、ジュニア債(中リスク)、株式(高リスク)とすることで、治療効果が低い場合には株式部分が損失吸収し、治療効果が高い場合には、製薬企業の利益が増加するという特徴を有します。このモデルをSMAの遺伝子治療薬であるゾルゲンスマを用いてシュミレーションをしており、その結果、ほぼ全ての想定において費用対効果が成立し、保険者、製薬企業、投資者の全てにとって有望なモデルであることが示されました。この研究は、超高額な遺伝子治療をどう支払うか、という課題を金融工学で解決しようとする研究で、高額な医療費を金融商品に変換するというユニークなアイデアが提案されています。簡単にいうと、住宅ローンの証券化を応用してみた、というお話しです。さて、日本でこれを実装することが可能かと言えば、いくつかの問題がありそうです。国が保険者で単一であることから、米国などより適応性が高いと考えられ、長期の追跡が容易である点も制度の運用に有利でしょう。一方で医療費の分割払いなど法制度の整備が必要であり、医療費の金融商品化の制度も整備されていません。また倫理社会的議論も必要でしょう。今回のモデル設定には、いくつか限界や問題点もあるので、すぐにこのまま導入することは難しいと思われますが、非常に魅力的な提案と思い、提示させていただきました。
文責:井上 健
DOI: https://doi.org/10.1038/s41434-026-00604-6
AAVLINK:Cre組換え酵素を用いた大型遺伝子導入カーゴによるAAV遺伝子治療の開発
AAVLINK: A potent DNA-recombination method for large cargo delivery in gene therapy
Jianbang Lin et al. Cell 2026:189, 1–18
今回は、新たなAAV遺伝子治療の技術開発に関する論文を紹介します。ご存知の通り、AAVを用いた遺伝子治療の大きな制約の1つが導入できる遺伝子のサイズが小さいことです。諸々含めて5kbほどが上限であり、これ以上の大きさの遺伝子をAAVに入れて細胞に送り届けることはできません。これまでこの問題点を克服するため、導入する大型の遺伝子を複数のピースに分けて2個以上のAAVに分散して搭載し、これを後から接着する技術としてDNA trans-splicing、RNA trans-splicingそしてprotein trans-splicingなどが開発されてきました。しかしこれらに技術に共通する欠点として、再構築された導入遺伝子産物の発現効率が著しく低く、治療薬としての実用化に耐えうるものではないことが挙げられます。本研究はこの問題を解決する新技術となります。筆者らはよく知られているCreリコンビナーゼというバクテリオファージ由来のタンパク質を用いています。ノックアウトマウスの作成では、おなじみのこの酵素ですが、loxPという短いDNA配列を認識して高い効率で正確に組み換えを誘導することができます。この研究では、AAVベクターにこのCreを分割した導入遺伝子とともに発現させ、高い効率で再構築することを可能にするのですが、肝はスプライシングドナーとアクセプターをうまく組み込むことで、Creの発現を一時的のみに限定することで、この外来性遺伝子産物の細胞への影響を最小限に止める工夫がされています。このシステムを使って彼らは実際にSHANK3やSCN1Aといった小児神経科ではおなじみの疾患遺伝子をAAVでマウス脳に導入することで、疾患モデル動物の表現型が改善することを確認しています。またマカクザルの脳への導入にも成功しました。ゲノム編集に用いるCas9の導入とゲノム編集(塩基編集を含む)も効率よく起こることが確認され、さらにヒト疾患遺伝子(多くはASD関連遺伝子など神経発達に関連する遺伝子)198個の遺伝子の導入ベクターを構築し、実際に細胞で全長のタンパク質を発現できることを確認しました。この中にはPOLR3関連白質変性症の責任遺伝子POLR3Aも含まれています。さらにCreを不安定化したAAVLINK2.0を作成し、安全性を高めたシステムも構築しております。今回の報告は、主に技術的な話ですが、5kbを超える大型の遺伝子導入を高い効率で行うことができる点で革新的です。今後、この技術を用いることで、これまで遺伝子サイズの問題から開発が躊躇されていた疾患についても治療法開発研究が進むことが期待されます。
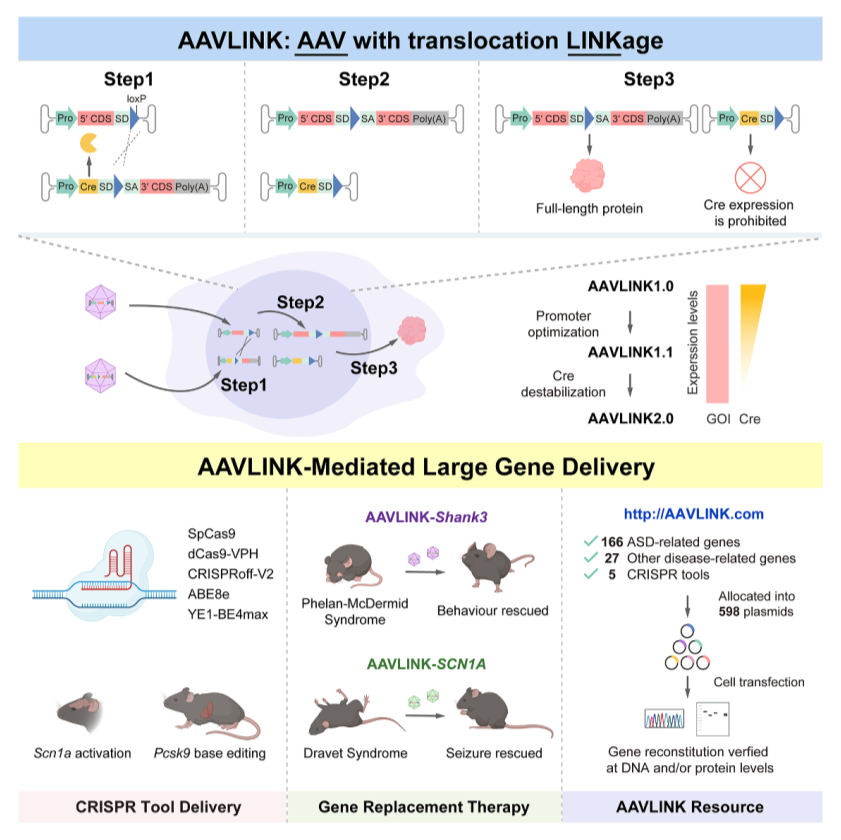
文責:井上 健
DOI: https://doi.org/10.1016/j.cell.2025.12.039
個別化遺伝子治療の時代へ:介入遺伝医学への次の一手
How to create personalized gene editing platforms: Next steps toward interventional genetics
R.C. Ahrens-Nicklas K. and K. Musunuru. Am J Hum Genet. 112, 1–4, 2025
前々月に紹介した論文の続きの話になります。論文というより、論説・状況報告に近いです。筆者らは、生後早期に50%の患者が死亡する尿素サイクル代謝異常症であるカルバミルリン酸合成酵素欠損症(carbamoyl-phosphate synthetase 1 (CPS1) deficiency)の症例に対して、出生後直ちに遺伝学的診断を行い、生後7ヶ月時に完全カスタムメイドのゲノム編集(塩基編集)治療を行なった症例について報告し、たった1例の超希少性疾患の患者さんに対して、診断からFDAの承認を得た治療までを超短時間で実施することができることを示しました。その後、どのようにこの試みを次の戦略につなげているか、というお話です。つまり「1人だけを救う治療を例外的に許可する」状況から、「個別化治療を標準医療に組み込む」仕組みへの転換が必要だと指摘しています。彼らはCRISPRを使った「プラットフォーム型治療」の仕組みを提案しています。CRISPR治療の利点は、そのプログラム性です。用いる遺伝子編集酵素を変えず、DNAの様々な変異を狙うためのガイドRNAの20塩基だけを差し替えることで、多くの患者ごとの変異に対応できるという「共通構造を持つ治療群(プラットフォーム)」が作れます。この発想を使えば、同じ病気でも変異が異なる多くの患者を、1つの大きな臨床試験(マスタープロトコル)にまとめて登録できる可能性があります。これを実現するためにFDA(米国規制当局)との議論を進めているわけですが、注目点はどこまで共通化できるのか、ということになります。彼らはフェニルケトン尿症(PKU)用プラットフォームと尿素サイクル異常症(UCD)用プラットフォームについて、検討を進めていますが、それぞれの変異や遺伝子について、非臨床評価を行うのではなく、同じLNP(脂質ナノ粒子)を使う限り、1種類の動物試験で複数の変異版をカバーできると認められました。追加変異への“迅速追加”も可能で、新たな患者変異を治療対象にする際、培養細胞での評価だけで試験への追加を認められる方向性が示されました。さらにUCDでは、7つの原因遺伝子すべてを統合する「umbrella of umbrellas(多層型傘)試験」案に基本的にFDAが同意し、患者が現れた遺伝子から順にIND(治験申請)を追加し、1つの巨大な臨床試験に統合していく設計が可能になりました。この仕組みが整えば、 1人のための“特例治療” から、多くの患者が恩恵を受ける“標準治療”への道筋ができます。どの変異でも迅速に 治験申請を追加し、同じ治療枠組みの中で評価することで、複数の患者のデータが蓄積され、承認が現実的になります。極めて珍しい遺伝病の患者にも、治療への道が開かれ、従来は製薬会社が参入しにくかった領域でも、共通プラットフォームがあれば持続可能な治療開発が可能になります。こういった取り組みにはNIHやARPA-Hなど米国の公的機関が既に積極的に資金を投じ、FDAも柔軟な制度作りを進めているとのことです。超希少疾患に対する遺伝子治療薬開発を実現するためのプラットフォーム型遺伝子編集治療の規制枠組みの整備を強く提言しています。
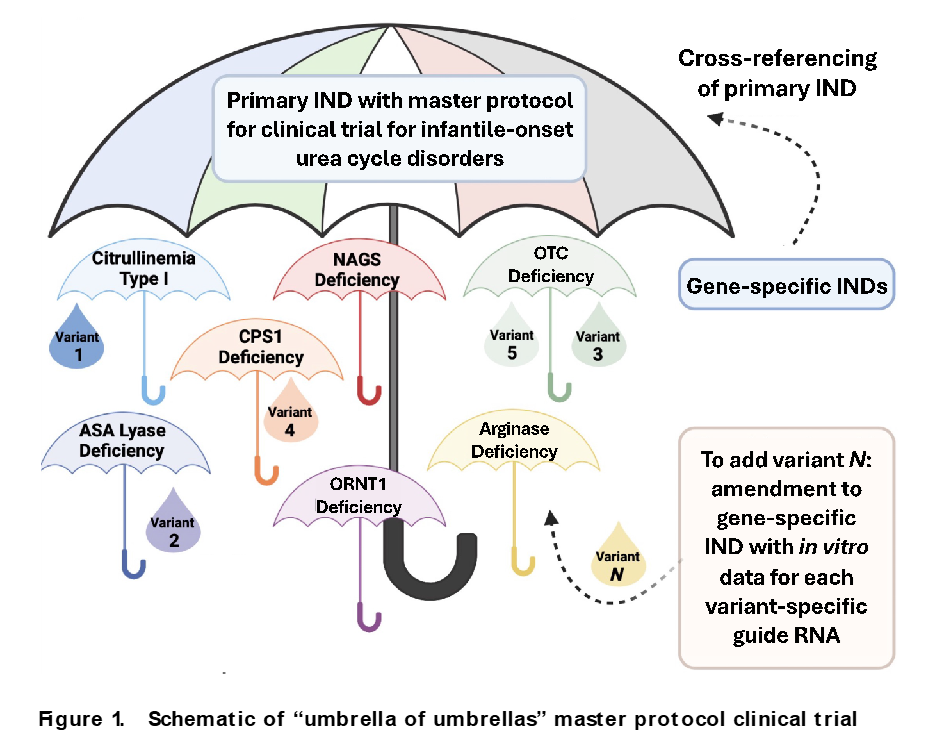
文責:井上 健
DOI: 10.1016/j.ajhg.2025.10.006
カナバン病患者に対するオリゴデンドロサイトを標的としたAAV遺伝子治療
Oligodendrocyte-targeted adeno-associated virus gene therapy for Canavan disease in children: a phase 1/2 trial
L. Leone et al. Nature Medicine
Nat Med. 2025 Sep 16. Online ahead of print.
米国の製薬企業Myrtelleが行っているカナバン病に対するアデノ随伴ウィルス(AAV)遺伝子治療臨床治験の途中経過に関する報告です。カナバン病は小児の遺伝性白質変性症の1つで、本研究班の対象疾患の1つです。多くの場合、出生直後は症状を認めませんが、数ヶ月で筋緊張低下、知的および運動発達の遅れ、大頭、てんかんなどを呈し、退行していく進行性の疾患です。ユダヤ系人種で頻度が高く、日本人では稀な疾患です。アスパルトアシラーゼ(ASPA)というオリゴデンドロサイト(髄鞘を作る細胞)に存在する酵素を作る遺伝子の異常で生ずる常染色体潜性遺伝疾患で、この酵素がないと正常な髄鞘を作ることができません。脳内にこの酵素が分解する基質であるNAAが溜まってしまうため、MRS検査(脳内の物質の濃度を測ることができるMRI検査)でNAAが上昇することが知られています。筆者らは、カナバン病の遺伝子治療を目指して、これまでもAAVにASPA遺伝子を組み込んで患者の脳に直接投与する臨床研究に取り組んできました。例えば2012年に公表された論文では、AAV2という血清型を用いた臨床研究で、NAAの低下を観察しましたが、臨床症状の改善までには至りませんでした。今回の論文では、Olig001というオリゴデンドロサイトに特異性が高い合成血清型を用いて、オリゴデンドロサイトを標的とした治療を行い、これまでより優れた治療効果が得られていることが注目点です。今回の報告は治療開始から1年後の途中経過の報告で、安全性の評価とともに治療効果についても報告されています。この臨床治験では、8人の乳児型カナバン病の患者さんに対して、脳外科的カテーテル挿入により脳室内にAAVが投与されました。外科処置に伴う副反応が数名で見られましたが、ウィルス投与による副反応は認めなかったようです。疾患自然歴を対照として治療効果についての検証を行ったところ、まず脳脊髄液のNAA濃度の速やかな低下を認めました。一方でMRS検査による脳内のNAA濃度は、現段階では有意な低下は見られていません。また合成MRIという手法を用いて髄鞘の体積を測定したところ、有意な増加が見られました。他にも言語運動知覚能力を評価できるMSELでは、いくつかのパラメータで有意な改善を認めました。今回用いられた新しい血清型は、従来のものに比べてオリゴデンドロサイトへの指向性が高く、髄鞘形成不全をきたす他の疾患の遺伝子治療法にも応用できるものであり、その点でも注目したい研究です。なおMyrtelle社はペリツェウス・メルツバッハ病の遺伝子治療にも取り組んでいることが知られています。
文責:井上 健
DOI: 10.1038/s41591-025-03919-w
出生直後の超希少性難病患者に対するカスタムメイド生体内ゲノム編集治療の試み
Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease
K. Musunuru et al. N Engl J Med 2025;392:2235-43.
今年の米国遺伝子細胞治療学会で大きな話題となった論文です。非常に重篤な経過をたどり、通常では生後早期に50%の患者が死亡する先天性代謝異常症であるカルバミルリン酸合成酵素欠損症(carbamoyl-phosphate synthetase 1 (CPS1) deficiency)の症例に対して、出生後直ちに遺伝学的診断を行い、生後7ヶ月時に完全カスタムメイドのゲノム編集(塩基編集)治療を行なった症例についての報告です。たった1例の超希少性疾患の患者さんに対して、診断からFDAの承認を得た治療までを超短時間で実施することができることを示した論文です。この疾患は、主に肝臓においてタンパク質の代謝によって生じるアンモニアを分解する第1段階の酵素をコードする遺伝子CPS1が欠損することで生ずる常染色体潜性遺伝の疾患で、生存することができても高アンモニア血症による重度の神経後遺症を残すことが知られています。筆者らは出生後直ちにゲノム解析にて診断をつけ、ゲノム編集治療薬の開発を開始する一方で、患者は対症療法による保存的治療を開始しています。その後の経過は、図1に記されたとおりですが、企業との協同でゲノム編集薬の設計開発(アデニン塩基編集ユニットをコードするmRNAと標的配列特異的なgRNAを脂質ナノ粒子LNP:Abe-kと命名)、患者変異を有するゲノム配列をレンチウィルスを用いて導入した肝臓系譜細胞の確立、同じ配列をRosa領域(遺伝子導入しても安全であることが知られている領域)に導入したマウスライン、これらを用いた治療効果の評価、オフターゲットの評価、安全性評価を次々に実現しながら前臨床試験を完遂し、並行してFDAとの折衝をすすめ、生後7ヶ月目に新薬臨床試験開始申請をおこない、1週間後に承認を得た後、直ちに1回目の投与が行われています。その後、臨床的評価をしつつ、効果不十分との判断の元、さらに1ヶ月後に2回目の投与が実施され、臨床指標の改善を観察したとのことです。今後、長期的な治療効果の検証が行われるとのことですが、近年注目されている製薬企業の通常の医薬品開発パスに乗らないような超希少遺伝性難病患者に対するパーソナライズド遺伝子治療についての実証例として、「やればできる」ことを証明した注目すべき症例報告だとおもいます。この研究には多くの企業がパートナーとして関わっており、こういったパーソナライズド分子治療の実証化に必要なチーム体制の重要性が示されていると思います。
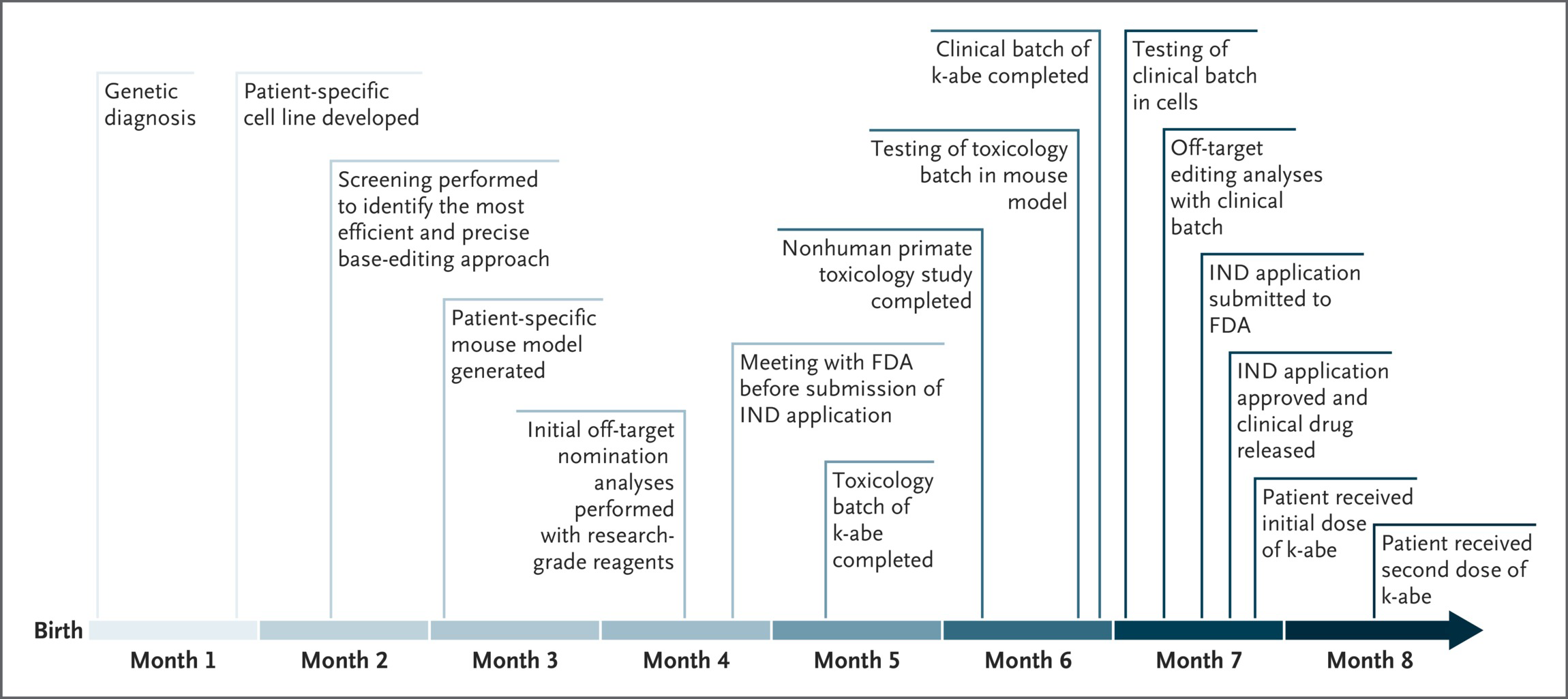
文責:井上 健
AAVを用いた脳血管関門通過型β-galactosidase発現はGM1ガングリオシドの蓄積を正常化した
AAV expression of a blood-brain barrier-penetrating form of b-galactosidase normalises GM1 ganglioside storage in mice
Saki Kondo Matsushima et al. J Clin Invest 2025 Apr 8:e180724.
最近、慈恵医大小児科から報告されたライソゾーム蓄積病の1つであるGM1 gangliosidosisに対する画期的な遺伝子治療法の開発に関する論文です。GM1 gangliosidosisはライソゾーム酵素の1つβ-galactosidase (GLB1)の欠損が原因で、その基質であるGM1が脳内に蓄積して起こる常染色体潜性遺伝疾患です。GM1 gangliosidosisに限らず、ライソゾーム蓄積病については、これまで酵素置換療法やex vivoおよびin vivo遺伝子治療など様々なプラットフォームでの治療法が開発され、いくつは臨床での実用化あるいは治験段階まで来ており、小児神経科領域での遺伝子治療開発におけるトップランナーの1つになっています。ライソゾーム酵素の補充療法において、1つの大きな特徴は、神経細胞をはじめとする体細胞は、細胞外に存在する酵素タンパク質をendocytosisで取り込んで利用することができる点で、これはcross-collection mechanismと呼ばれており、治療においては必ずしも標的細胞全てに対して直接補充をしなくても良いことが知られています。一方で酵素置換療法など末梢循環に投与された酵素は、脳血管関門(BBB)を通過することができないため、中枢神経症状を改善させることは困難でした。近年、BBB通過能を持つモダリティの開発が進んでいますが、今回用いられたトランスフェリン受容体(TfR)は近年特に注目度の高い分子です。具体的にはTfRに結合する抗体の可変部位のペプチド配列をコードするDNA配列を補充するタンパク質(本研究の場合はGLB1)のcDNAに結合させたものをAAVに組み込みます(図を参照)。今回は、あえて肝臓特異的に発現させるプロモーターを用いて、肝臓でBBB透過能を持つ修飾GLB1を発現させ、その中枢神経系への移行と治療効果の評価について、GM1 gangliosidosisモデルマウスを用いて行っています。実験は、生後10週目に静脈内投与で行われています。治療効果は劇的で、十分な血中酵素濃度、中枢神経系への移行、脳でのGM1蓄積の改善、寿命の延長が確認されております。一方で行動実験による神経学的臨床症状については、改善は一部にとどまっており、これは投与を1ヶ月早くしても変わらなかったとのことなので、この点は今後の臨床応用における懸念点の1つになるかもしれません。現在、AAVを用いた中枢神経系への脳室内投与によるGLB1補充療法が開発されておりますが、本研究で用いられた手法は静脈内投与が可能である点で大きなメリットがあります。
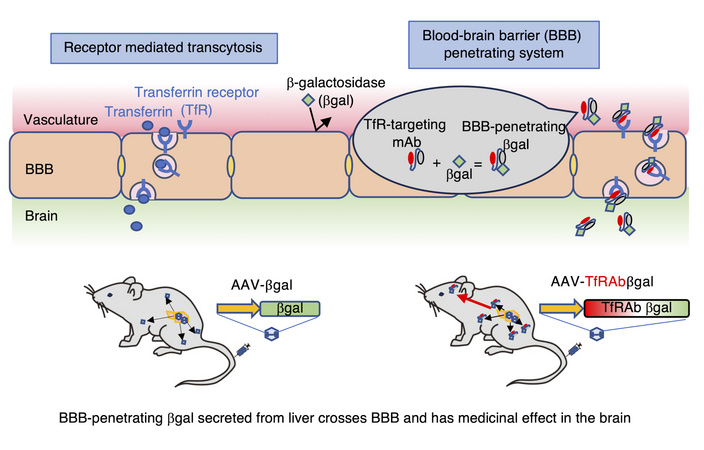
文責:井上 健
https://doi.org/10.1172/JCI180724
皮質下嚢胞をもつ大頭型白質脳症モデルマウスに対する遺伝子治療は脳浮腫と運動機能を改善させる
Gene therapy rescues brain edema and motor function in a mouse model of megalencephalic leukoencephalopathy with subcortical cysts
Alejandro Brao et al. Molecular Therapy (2025)
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) は一定年齢までは正常に発達するにもかかわらず、後に進行性に大脳白質障害を来し、徐々に退行する進行性白質脳症の1つです。診断は特徴的なMRI所見と臨床症状、そして遺伝子解析によって行われます。これまでに4つの原因遺伝子が知られていますが、最も頻度が高いのは、MLC1の両アレルに起こる機能消失型変異で、常染色体潜性遺伝形式をとる極めて稀な疾患です。MLC1はグリア細胞の1つであるアストロサイトで発現しており、本疾患はアレクサンダー病や白質消失病とともにアストロサイトの機能不全に起因する疾患であることが知られておりますが、どの疾患もまだ根治療法はありません。今回紹介する論文では、ヒトMLC1遺伝子をAAVを用いて導入する遺伝子補充治療法開発の試みです。筆者らは、以前の報告で、アストロサイト特異的発現を可能にするGFAPプロモーター下にhMLC1cDNAを配置し、これをAAVrh10という血清型を用いて、Mlc1ノックアウトマウスの小脳のくも膜下腔に直接投与したところ、脳の空胞化を含む症状を改善させることができました。しかし実際の患者への投与を考慮した場合には、AAVを全脳に行き渡らせる必要があることがわかりました。そこで、本研究では、脳血管関門(BBB)透過型合成血清型であるAAV9P31を用いた静脈からの全身投与による治療効果の検証を行なっています。本治療法は、先行研究でも明らかとなっているのですが、すでに脳病変と症状が出現している個体への投与でその症状を改善させることができる点が特筆に値すると思われます。今回の報告では、生後10ヶ月齢の有症状のMlc1ノックアウトマウスにAAVを静脈内投与し、22カ月齢で解析しています。その結果、AAVは広く脳全体に行き渡り、ほとんどの場所で生理学的発現レベル以上のMLC1の発現を可能にしています。GlialCAMの機能的発現回復も見られ、白質の空胞化も消失しています。MRI画像では、T2強調画像や拡散画像で脳の水分量がほぼ正常化し、脳浮腫の所見がほぼ消失。行動試験でも投与前に見られた異常所見が、治療後に改善しております。これらの結果は、以前の彼らの報告をさらに一歩、臨床実用化、特に静脈内投与による非侵襲的な遺伝子治療の実用化に近づけるものとして、注目に値すると考えました。
文責:井上 健
https://doi.org/10.1016/j.ymthe.2025.02.046
塩基編集治療はヒト化プリオン病マウスモデルの寿命を延長させる
In vivo base editing extends lifespan of a humanized mouse model of prion disease
An et al. Nature Medicine (2025)
今回はクロイツフェルト・ヤコブ病に代表されるプリオン病に対する遺伝子治療法の開発に関する論文を紹介します。プリオン病は主に中高年が罹患し、急速に脳のスポンジ様変性が進む難病中の難病です。85%が弧発型、15%がプリオン遺伝子(PRNP)の変異による遺伝性で、少数で感染による症例が知られていますが、以前狂牛病として世界中で話題になりました。プリオン病は、構造変化をきたしたPRNPタンパク質が脳の中に広がっていく、あるいは感染することが原因で起こることが知られており、疾患の進行や感染の成立のためには宿主のプリオンタンパク質の存在が必要であることから、プリオン遺伝子を破壊することが感染防御や治療のために有効であることが知られています。マウスではPrnp KOは表現型を呈さず、人ではheteroでLOFが一般集団に存在することから、少なくともハプロ不全での異常は起こさないと考えられます。現在、アンチセンスオリゴによる治療薬が治験に入っていますが、複数回投与が必要であるこの治療法のデメリットを回避できる遺伝子治療法の開発は有用と考えられています。筆者らは、今回、塩基編集(Base Editing; BE)技術を用いて、PRNPにナンセンスコドンを導入することで、この遺伝子を破壊することができる遺伝子治療法の開発を行いました(図1)。用いた塩基編集はBE3.9Maxという研究開始当時の最新型のものですが、その後、開発された改良品の検討も行っています。選び抜いたsgRNAはR37Xの早期終止コドンを導入するもので、HEK293T細胞を用いたin vitroで54%の効率で変異導入に成功。spCas9を用いたBEコンストラクトはサイズが大きく、1つのAAVには載らないため、inteinを用いて2つに分割したBEを別々のAAVに搭載したdual AAVシステムで構築し、脳血管関門透過性を有する血清型であるPHP.eBを用いて野生型マウスに眼窩静脈内投与しています。その結果、35日後に20%の変異導入効率、600日後には37%まで効率が上昇しています。次にこのAAVを用いてプリオン病のモデルマウスへの投与実験を行っています。これはPrnp ノックアウトマウスにヒトPRNP遺伝子を含むBAC トランスジェニックマウスを掛け合わせたヒト化マウスで、AAV投与後にCJD(弧発例とE200K変異例)の脳抽出物を投与し、寿命を観察しました。安全性を考慮し、脳抽出物の投与後には脳を用いた解析は行なっていません。その結果、弧発CJD脳抽出物投与群では、BE AAV投与により59%の寿命延長効果が見られ、遺伝型CJD脳抽出物投与群では、BE AAV投与により44%の寿命延長効果が見られました。その後、筆者らはより効率の良いBEやsgRNAなどの改良を加え、AAV投与量を6分の1以下に減らすことに成功し、大量投与に伴う副反応の発生を抑えることにも成功しています。プロモーターの最適化、組織特異的miRNA標的配列の挿入、off-target効果など安全性を高める改良や検証も行い、全体として即臨床応用が可能なレベルにまで仕上げている点は印象的です。塩基編集によるゲノム編集遺伝子治療は、BEの改良が進み、臨床応用が極めて近いレベルにきていることを示すスタディだと感じました。問題点としては、最大効率でも50%に満たない編集効率であるため、進行を遅らせることはできても止めることはできない点、つまり治療のパワーはまだ十分ではない、ということです。とは言え、この点は今後の技術開発により解決できる要素なので、今回の非臨床POCが得られた点は大きな進歩であると言えます。同様のプラットフォームは、PMD、H-ABC(TUBB4A)、アレクサンダー病などでの応用が可能でしょう。
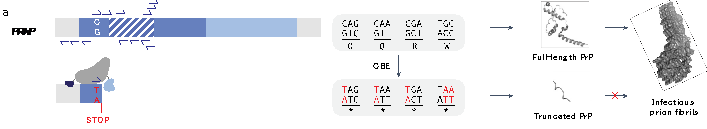
文責:井上 健
https://doi.org/10.1038/s41591-024-03466-w
Tubb4a発現抑制によるH-ABC白質変性症の治療
Therapeutic suppression of Tubb4a rescues H-ABC leukodystrophy.
Sunetra Sase et al. bioRxiv (2024)
今回はpeer review前の論文の紹介となります。基底核および小脳萎縮を伴う髄鞘形成不全症(H-ABC)は、先天性大脳白質形成不全症の1つで、細胞骨格を構成するタンパク質の1つであるチュブリンをコードする遺伝子TUBB4Aの変異が原因で起こる稀な疾患です。臨床的には生下時には異常なく、通常の発達過程を呈しますが、徐々に発達が退行し始め、痙性、小脳失調、錐体外路症状を呈する疾患です。本疾患は、TUBB4Aに新生突然変異で生ずるアミノ酸置換変異が原因となることが多く、特にp.Asp249Asnの変異が多いことが知られています。MRIでは、髄鞘形成不全に加え、特徴的な基底核と小脳の萎縮を認めます。本論文では、このH-ABCの治療法を開発するために、TUBB4Aの発現を抑制するアンチセンスオリゴ(ASO)を開発し、その有効性について疾患モデルマウスを用いて検証しております。まず著者らは、元々報告していたp.Asp249Asn変異を組み込んだマウスとTUBB4Aのノックアウトマウスを掛け合わせて、適切な疾患モデルとなりうるマウスを探索します。その結果、片方の染色体のTUBB4Aにp.Asp249Asn変異をもち、もう片方の染色体のTUBB4Aは欠損しているマウスが最も適していることを見出しました。なおTUBB4Aの完全欠失は、マウスにおいて全く症状を引き起こさないことが知られています。生直後のマウス(Tubb4aD294N/KO)に、TUBB4Aを標的としてデザインされたASOを脳室内に投与しました。その結果、通常100日前後で死亡するマウスが300日を超えて生存し(図1)、運動機能や髄鞘タンパク質の発現、髄鞘形成、電気生理学的所見(VEP)などの指標が全て著明に改善することが示されました。これらの結果から、ASOを用いたTUBB4Aの遺伝子発現抑制治療は、H-ABCの根治療法になりうることが示されました。今後の臨床応用が期待されるところです。先天性大脳白質形成不全症の治療薬の開発が徐々に進み、これらの疾患の根本的な治療が議論される時代となりました。診断から治療への大きな転換点に差し掛かっています。

文責:井上 健
https://doi.org/10.1101/2024.08.27.609903
白質消失病のマウスモデルに対するAAV遺伝子治療の安全性と治療効果に関する検討
Evaluation of safety and early efficacy of AAV gene therapy in mouse models of vanishing white matter disease
Jessica A. Herstine et al. Molecular Therapy 15:7259 (2024)
白質消失病Vanishing white matter (VWM)は、進行性白質脳症の1つで、1から5まであるEIF2Bのサブタイプ遺伝子(EIF2B1~5)におけるホモあるいは複合ヘテロ変異による常染色体潜性遺伝を示す疾患である。現在までに有効な治療法はない。本論文は、この疾患に対するAAVを用いた遺伝子治療法開発の報告である。本論文では、EIF2B複合体を構成する構成タンパク質の1つ、EIF2B5に点変異を持ち、重症度の異なる2系統のマウスモデルが用いられている。以前、これらのマウスを用いた基礎的研究において、VWMはグリア細胞の1つであるアストロサイトにおけるEIF2Bの機能不全が原因であることが明らかになっている。そこで筆者らは、アストロサイト特異的に発現するGFAP遺伝子のプロモーター配列を用いて遺伝子治療用のベクターを構築し、これを用いた治療を行い、その効果を検証している。AAVの血清型はAAV9を用い、アストロサイト特異的発現とAAVへのパッケージングを可能にした独自の短縮型のGFAPプロモーターを構築している。補充する遺伝子はヒト型EIF2B5である。AAVは、出生後1日目に脳室内投与されている。この短縮型のGFAPプロモーターを用いたAAVは、2系統のマウスどちらについても、汎用されるCMVプロモーター、サイズオーバーの通常型GFAPプロモーターを用いたAAVよりも有意に高い治療効果が示された。すなわち体重や寿命などの一般状態、行動試験による運動機能、MRIによる白質病変についての改善が示されている。本研究により、VWMに対する遺伝子補充療法の基盤となるプラットフォームを作ることができたと考えらえる。一方で症状の改善は完全とは言えず、治療効果は十分ではない。また今回はなぜか、治療効果の組織学的な検討の所見が示されておらず、まだそのまま臨床応用が可能なレベルには至っていない印象である。今後のさらなる改善が期待される。
文責:井上 健
https://doi.org/10.1016/j.ymthe.2024.03.034
痙性対麻痺50型に対するAAV遺伝子治療:患者1人への第1相治験
AAV gene therapy for hereditary spastic paraplegia type 50: a phase 1 trial in a single patient
James J. Dowling et al. Nature Medicine 30 :882–1887 (2024)
希少性難病は1万種以上の疾患が知られているが、ほとんどの疾患には治療法が存在せず、その治療法開発は社会的なデマンドが大きい。遺伝子治療/ゲノム編集技術等の進歩によりこれらの疾患の治療法は概念的には可能となってきたが、患者数が少ない、開発コストが過大である、利益が得られない、などの理由によりその開発は進んでいない。この論文で筆者らは、カナダのSick Children病院において診断された1名の痙性対麻痺50型患者に対して、個別医療として遺伝子治療の試みを成功させた事例を報告している。この患者はカナダで唯一の痙性対麻痺50型患者で、18ヶ月齢で全エクソーム解析にて遺伝学診断が確定した。診断後間も無く患者家族が本疾患の遺伝子治療を確立することを目的としてCureSPG50という財団を設立した。この財団が核となり、複数の施設の共同研究チームにより、疾患原因遺伝子AP4M1を補充するためのアデノ随伴ウィルス(AAV9-AP4M1)を作成し、非臨床試験を経て、髄腔内投与により診断から3年以内となる4歳時に対象患者1名のみの第1相治験が実施された。投与量は1 × 1015 vector genomesでかなりの高容量である。また免疫副反応と抗AP4M1抗体の産生を防ぐために3種類の免疫抑制剤を用いた濃厚な免疫抑制治療が行われた。主要評価項目は安全性と忍容性、二次評価項目として治療効果が検証された。治療開始後、12ヶ月時点で忍容性は良好であり、重篤な副反応はみられず、一時的な軽度の副反応が見られたのみで、これらも治療により軽快した。治療効果の評価については、疾患自然歴と比較した今後の長期的な解析を待つ必要があるが、現段階では症状の安定化が観察されている。本報告では、希少性難病に対する個別化遺伝子治療の開発についての実例を提示した。この実例を通じて、様々な問題と解決策が明らかになったが、費用面では非臨床での開発に3億円以上、臨床治験の実施に2千万円以上の費用が必要となっている。今後、多くの事例に適用するには費用面での大幅な軽減策が必要であることも明らかとなっている。
文責:井上 健
https://doi.org/10.1038/s41591-024-03078-4
遺伝子量を補正できる合成miRNAサーキットは精度の高いRett症候群の遺伝子治療を可能にする
Synthetic dosage-compensating miRNA circuits allow precision gene therapy for Rett syndrome
M. J. Flynn et al. bioRxiv. 2024 Mar 14:2024.03.13.584179.
多くの中枢神経系の疾患の原因遺伝子は、その遺伝子発現量が高い精度で制御されていることが多い。そのため、疾患遺伝子の欠損のみならず、重複などによる過剰発現も疾患を引き起こすことが稀ではない。例えばRett症候群はX染色体上に位置するMECP2遺伝子の欠損で起こる女児に特異的な疾患であるが、同じ遺伝子の重複は男児における知的障害症候群を引き起こす。PLP1も然り、欠失も重複も疾患の原因となる。従って欠失症候群に対して、通常の遺伝子治療による補充療法は、過剰発現による副反応を引き起こす危険性が高い。この問題は、多くの遺伝性疾患における遺伝子治療の実用化の障壁となっている。つまりAAVによる遺伝子治療法は、1つ1つの細胞レベルでは様々な導入コピー数に起因する発現多様性が生ずるからである。もしこれらの変動レベルを一定に保つことができれば、この問題に対する解決につながるが、本論文は、この解決法の1つを提案するものである。筆者らは人工的なマイクロRNAを用いたincoherent feed-forward loop circuit(IFFL;無秩序前向き送りループ回路)という方法を用いて細胞や脳の中でMECP2の発現量を正確に制御する手法を確立し、これを組み込んだAAVを用いてRett症候群モデルマウスに対する治療効果を改善することに成功した。簡単にいうと、これは目的遺伝子発現ユニットの下流の3‘非翻訳領域内に設計したイントロンの中に人工的なmiRNA発現ユニットを設置し、そのすぐ上流の3‘非翻訳領域にmiRNA標的配列を組み込むことで、ベクターからの導入遺伝子の過剰発現を抑制的に制御する手法である。この発現制御回路を導入したマウス脳を1細胞レベルで内在性と外来性のMEPC2mRNAを解析したところ、様々な量で遺伝子が導入されているにもかかわらず、一定のレベルに正確に制御され、しかも持続的な遺伝子発現が維持されていた。全身投与で脳特異的な遺伝子治療ができる血清型を用いたRett症候群モデルマウスに対するAAV遺伝子治療では、IFFLを用いて発現制御した治療は、制御されていない治療に比べ行動面での症状を24週間に渡って大きく改善させることができた。これらの結果より人工的なマイクロRNAを用いた遺伝子発現制御回路は生体内での正確な遺伝子発現を制御し、遺伝子治療の安全性と治療効果を高めることができることを示した。
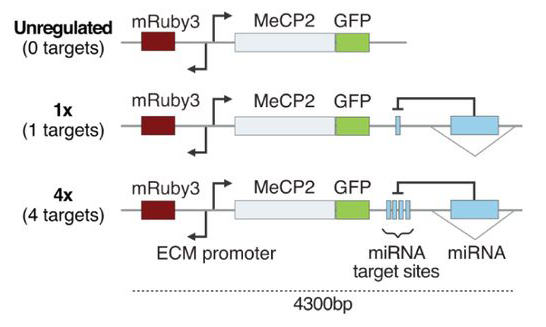
文責:井上 健
doi: 10.1101/2024.03.13.584179.
ヒト型トランスフェリン受容体に特異的に結合する合成AAVは脳への高いデリバリー能を有する
An AAV capsid reprogrammed to bind human Transferrin Receptor mediates brain-wide gene delivery
Qin Huang et al. bioRxiv preprint [Preprint]. 2023.
今回はまだ正式に公表されていない論文の紹介になります。AAVの血清型は自然界に存在するものに加え、様々な手法でAAVのcapsid(ウィルスの外箱を構成するタンパク質)のDNA配列に変化を加えることで、自然界にあるAAVよりも特異性が高く治療効率も良い合成AAVを作り出すことが可能です。血管内投与されたAAVが血管脳関門(BBB)を超えて脳内に効率よく入ることができるためには、BBBを構成する血管内皮細胞を通り抜けることが必要ですが、これまでどのような受容体を介してBBBを通過するのかわからないまま、いわばめくら打ちで合成AAVが作られていました。今回の研究論文ではヒトの血管内皮細胞に発現していることがわかっているトランスフェリン受容体(TfR1)に結合する合成AAVをin vitroの系で選択し、これを実際に細胞や動物を使って高効率にBBBを通過することができることを証明しております。BI-hTFR1は、AAV9のVP1配列に7残基の挿入させたライブラリーの中からヒトTfR1に特異的に結合する血清型を抽出することで得られました。さらに細胞で作られたBBBを効率よく通過することを確認した後、マウスに血管内投与されたBI-hTFR1が脳内の細胞に効率よく感染することを確かめています。この際、用いたマウスはヒト型TfR1を発現するように操作されたもの(TFRC KIマウス)で、ヒト型TfR1を発現しない野生型のマウスに比べ、40−50倍の感染効率の上昇が確認されました。感染した細胞は主に神経細胞とアストログリア細胞でした。オリゴデンドロサイトへの感染は確認されておりませんが、これはオリゴデンドロサイトでの発現が悪いCMVプロモーターを用いているからかもしれません。最後に彼らはゴーシェ病の原因遺伝子GBA1を組み込んだBI-hTFR1のAAVを合成し、AAV9との比較投与実験を行い、AAV9と比較して30倍のベクターを脳内に送り込むことができることを明らかにしました。BI-hTFR1の利点は、BBB通過の機序が設計の段階から明確であることから、治療効果の推測などが可能であること、マウスとヒトとのBBB透過性の違いによる治療効果の違いの影響を排除することができるため、マウスからヒトへのトランジションがスムーズになることなどが挙げられます。またヒトでの高いBBB透過性は治療に必要なAAV量を少なくすることを可能とするため、肝障害や免疫反応などの副反応を低下させることができると期待されます。
文責:井上 健
https://doi.org/10.1101/2023.12.20.572615
広範なPMA配列適合性を持つ小さなCjCas9バリアントの開発
Continuous directed evolution of a compact CjCas9 variant with broad PAM compatibility
L. Schmidheini et al. Nature Chem Biol 2023
今回も前回、前々回に引き続き、テクニカルな論文の紹介となることをお許し頂きたい。次世代の遺伝子治療プラットフォームになるであろうゲノム編集技術を、どうやってキャパシティが限られるAAVベクターに載せるか、という点に関する技術開発研究の紹介をしてきた。今回も同様のテーマであるが、これまでと異なる点は、ゲノム編集技術の本体であるCas9タンパクそのものに関する研究である。よく用いられる化膿性連鎖球菌由来のSpCas9は、cDNA長が4.1kbもあるため、ゲノム編集ユニットを単一のAAVベクター(キャパシティは全長5kb)に組み込むことが不可能である。一方カンピロバクター・ジェジュニ由来のCjCas9は、全長3kbを切る最短の長さであることから、遺伝子治療への応用が期待されている。反面、PAM配列が長いため、sgRNAが設定できる場所(つまり編集を行うことができる部位)が限定されること、活性がSpCas9に比べて低いことなどが問題点であった。そこで本研究では、ファージと大腸菌を用いたシステム(PACEと名付けられている)を用いて、これらの課題を克服した新たなCjCas9バリアントを開発し、実際にこれをAAVベクターに組み込んで、各種細胞や動物モデルでその有用性を検証しているので紹介する。
ここではPACEの詳細は割愛するが、要は大腸菌の中でファージが生き残るためのプレッシャーをかけ、生き残こるために必要な変異をファージに搭載したCjCas9に誘導することで、より緩やかなPAM配列と高い活性を持たせる4つのアミノ酸置換変異を進化的に作り出すことができた。これらに加え、以前から知られていたDNA切断活性の増強効果のある変異1つを導入したものがevoCjCas9である。evoCjCas9のPAM配列はN4AHおよびN5HAで、野生型のN3VRYACに比べて10倍の頻度でゲノム中に存在する。またDNA切断活性もSpCas9に迫るが、一方でオフターゲット効果は野生型CjCas9よりも高いことから、特異性については臨床応用という点での課題が残った。次にDNA切断活性を除去した不活型evoCjCas9に脱アミノ化酵素(デアミナーゼ)を付加した一塩基編集(base editing)や逆転写酵素を付加したプライム編集などを用いた修飾ゲノム編集治療ユニットを作成し、標的とする一塩基置換を誘導することができるか、細胞やマウスを用いた検証を行なった。これら修飾ゲノム編集は、DNAを切断する代わりに修飾を行うもので、オフターゲット効果によるDNA損傷の危険が少なく、疾患原因変異の修復が可能であるため、臨床応用が期待される技術である。特筆すべきは、AAVに搭載したevoCjCas9は、マウスの肝臓や脳においても、30−40%という高い効率で一塩基編集が可能であった点であり、治療効果という点では、すでに実用化可能なレベルに達していると言えるであろう。
前回、紹介したdual AAVシステムでは到達し得ない高い編集効率を得ることができ、高い標的設計自由度を持つevoCjCas9は、ゲノム編集とAAV遺伝子治療の組み合わせによる治療プラットフォームの開発における大きな進歩と言って良いであろう。
文責:井上 健
https://doi.org/10.1038/s41589-023-01427-x
mRNAトランス・スプラシングAAVベクターによる(エピ)ゲノム編集と遺伝子治療
mRNA trans-splicing dual AAV vectors for (epi)genome editing and gene therapy
L. M. Riedmayr et al. Nature Communications 2023; 14:6578.
前回に引き続き、今回もdual AAV vectorシステムを用いてゲノム編集モジュールを2つに分離し、これをin vivoで標的細胞に送り届けることによって可能になる新たな遺伝子治療のプラットフォームに関する研究を紹介する。AAVベクターは安全性と有効性が極めて高い遺伝子治療プラットフォームであるが、搭載できるDNA断片のサイズが小さいことが新規技術の開発や治療対象疾患の拡大に関する大きな障壁となっている。これを解決する1つの手法が、搭載したいcDNAを2つ以上の複数のDNA断片に分けてAAVに搭載し、感染した宿主細胞の中で再構築させるというものである。前回紹介した論文の中で使われていたものがsplit-inteinシステム(タンパク質ベースの再構築システム)であるのに対して、今回用いられている手法はmRNAレベルでの再構築を行うものである。筆者らがREVeRTと命名したこのシステムは、トランス・スプラシングと呼ばれる機構を用いてその再構築を行う。これは2つに分けたmRNAの3‘端にスプライシンドナー配列あるいはスプライシングアクセプター配列を組み込み、その先に2つのmRNA断片を対合させる相補的塩基配列を組み込むことによって2つのmRNA断片の間でトランス・スプライシングを誘導することで全長mRNAを再構築するシステムである(図参照)。
筆者らは、まずこのシステムが細胞内で機能するかについて、培養細胞を用いた系で検証・最適化を行った後、AAVに組み込んで様々な手法で生体内での検証を行った。例えばルシフェラーゼレポーターシステムを組み込み、マウス腹腔内注射により、心臓や脳を含む全身臓器での遺伝子再構築後の発現誘導を確認した。単一AAVによる導入効率の約20%程度の再構築効率を達成しており、実用に耐えうるレベルと考えて良いであろう。他にゲノム編集を応用したdCas9-VPRを用いた特異的遺伝子活性化モジュールを組み込み、マウスの目の網膜細胞に投与するUsher症候群に対する遺伝子治療モデル、ゲノム編集による遺伝子破壊と別遺伝子の活性化を並行して同時に行うCas9-VPRを用いた網膜色素変性症の遺伝子治療モデル、さらに6.8kbにおよぶ原因遺伝子cDNAを導入するStargardt病の遺伝子治療モデルなど様々な応用例(主に眼科領域)において、その有効性の検証を行なっている。split-inteinシステムとの比較も行い、同等レベルの有効性があることも報告されている。REVeRTの利点として、cDNAの2分割の設計の自由度が高いこと、再構築の効率がタンパク質の高次構造の影響を受けないこと、バレード由来のポリペプチドの導入による免疫反応のリスクを考慮しなくて良いことが挙げられている。
今後、先天性大脳白質形成不全症を始め、様々な疾患の遺伝子治療プラットフォームの設計を検討する場合、前回および今回の報告に示されたdual AAVシステムの使用は必要不可欠になってくると推測される。こういった技術の組み合わせによってAAVを用いた遺伝子治療の治療効果を高めることが可能になると期待される。
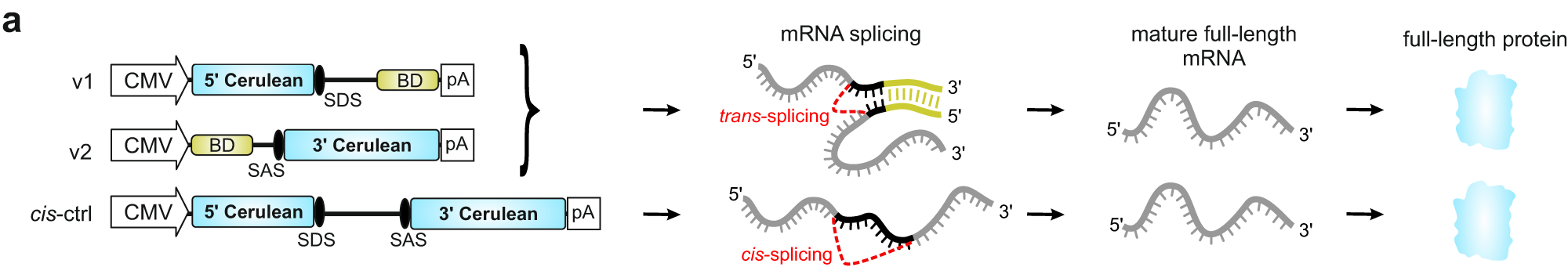
文責:井上 健
https://doi.org/10.1038/s41467-023-42386-0
AAVを用いた塩基編集治療による網膜色素変性症モデルマウスの治療
AAV-mediated base-editing therapy ameliorates the disease phenotypes in a mouse model of retinitis pigmentosa
Wu Y et al. Nature Communications 2023;14:4923.
多くの劣性(潜性)遺伝子形式をとる疾患に対する画期的な治療法として、欠損した野生型遺伝子を補充する遺伝子補充療法の臨床応用が進んでいる。しかし実際には、遺伝子の多くは、生理的に様々な発現制御を受け、非常にナイーブな遺伝子発現の調節がされていることが知られている。例えば細胞および時期特異的発現や選択的スプライシングなどがこれに該当するが、遺伝子補充療法では、こういった生理的な微細な制御の多くを無視し、欠損遺伝子を過剰発現するため、必ずしも治療効果が十分ではない、あるいは望まぬ副反応を呈することも多い。また優性(顕性)遺伝形式の疾患では、遺伝子補充療法そのものの効果が望めないことが多い。こういった問題を解決する1つの方法が、Cas9を応用した塩基編集技術である。これは二本鎖DNA切断機能を欠落させたdCas9に塩基編集酵素を結合し、標的部位の変異を野生型塩基に変換する(例えばAをGに変える)ことで治療を試みる次世代型ゲノム編集治療である。本報告では、外科的に患部に直接アクセスできる網膜をモデルとし、すでに遺伝子補充療法が試みられているPDE6B変異による網膜色素変性症のモデルマウスrd10に対して、AAVを用いた塩基編集遺伝子治療を試みている。
AAVは、神経系の多くの細胞がそうである非分裂細胞への導入効率が良い点、核へのインテグレーションが無いため、ガン化のリスクが低い点など多くの利点を持つ一方で、導入できる遺伝子のサイズが4.7kbに制限される。今回の報告では、塩基編集遺伝子SpRY-ABE8eのcDNAをN末とC末の2つに分け、それぞれを別のAAVに組み込み、別のタンパク質として発現させたのち、split-intein mechanismを用いて、細胞内でつなぎ合わせるdual-AAVシステムを用いることで全長のSpRY-ABE8eを発現させることに成功している。このAAVを生後14日のrd10の網膜に直接投与し、生後35日で各種評価を行ったところ、最高で49%(平均34%)の効率で標的の塩基編集に成功することができた。また非標的塩基の編集や標的部位のindelなどは12%ほどの効率であった。表現型解析では、PDE6Bタンパク質の復元、視細胞の形態の回復、電気生理学的な視機能の回復、視覚依存的な行動の改善などが示され、本遺伝子治療法の有用性が示された。
PMDを始め、多くの先天性大脳白質形成不全症は、単純な遺伝子補充のみでは治療が困難であることが推測される。本報告に示された塩基編集治療は、ゲノム編集とAAV遺伝子治療を組み合わせることで、まさに「元に戻す」根本的な遺伝子治療を治療レベルで可能にすることができる技術として、今後の応用が期待される。
文責:井上 健
https://doi.org/10.1038/s41467-023-40655-6
経頭蓋集束超音波照射による血液脳関門開放術:特定の脳領域への選択的アデノ随伴ウイルスベクター導入
BBB opening with focused ultrasound in nonhuman primates and Parkinson’s disease patients: Targeted AAV vector delivery and PET imaging
Blesa J et al. Science Advances 2023.
中枢神経系の疾患に対する遺伝子治療法開発における大きな障壁は、脳血管関門(BBB)の存在である。BBBは血管内皮細胞の密着結合(tight junction)による密閉構造による。BBBは血液中の細菌やウィルスなどの異物が容易に脳内に入らないように保護する働きをしており、このため血管内に投与した薬物や遺伝子治療用のAAVウィルスを脳内に到達させることは容易ではない。本研究では、これまで齧歯類で基礎的開発が行われてきたマイクロバブルと経頭蓋集束超音波照射の併用によるBBBの可逆的な解放術をサルとヒト患者に対して実施し、遺伝子治療用のAAVウィルスを脳内に安全に送り届けることができることを示した。
経頭蓋集束超音波照射はMRIガイド下に脳の特定領域に焦点を当てて超音波照射できる装置である。血管内にマイクロバブル(小さな膜球体にガスを閉じ込めた気泡で、超音波診断の造影剤としても用いられている)を投与し、低出力で経頭蓋集束超音波照射すると非侵襲的にBBBを一時的に開くことができる。筆者らは6匹のカニクイザルを用いて、この技術が安全かつ効果的に脳の狙った部位のBBBを選択的に開き、その部位に静脈内から投与したAAVが到達していることを明らかにした。すなわちMRIにおける血管造影剤として用いられ、BBBを通過しないことが知られているガドリニウムが、超音波照射により脳内に漏出し、MRI画像で増強効果を示した。また、この効果は照射後30日後には消失し、組織学的解析でも脳損傷やそれに伴うミクログリアの活性化やアストログリオーシスなどの変化もほとんど認めなかった。同時に血管内投与されたAAVは、ガドリニウムが漏出した部位に比べて限定的ではあるものの、脳内へのデリバリーが成立していることが明らかとなった。これらの知見を元に、さらに3名のヒトパーキンソン病患者に対し、臨床治験の一部として本法を用いたところ、明らかな副作用もなく、ガドリニウムの増強効果、さらに18F-Cholineをトレーサーとして用いたPETでも標的部位でBBBを可逆的に解放させることができることを示した。
本研究では、脳の限定的な場所に対して選択的にAAVを送り届けることができる技術の実証を行なったが、脳全体へのデリバリー効率を上げるために、この技術をどう応用していくことができるのかは、今後の課題となる。本研究には京都大学のグループが参加しており、今後我が国でも臨床応用を目指した技術開発が進むことが期待される。
文責:井上 健
https://doi.org/10.1126/sciadv.adf4888
単回全身投与によるARSAノックアウト異染性白質ジストロフィーモデルマウスに対する遺伝子治療
Single Systemic Administration of a Gene Therapy Leading to Disease Treatment in Metachromatic Leukodystrophy Arsa Knockout Mice
St. Martin T et al. J. Neurosci 2023 in press.
異染性白質ジストロフィー(MLD)は代表的な白質変性症の1つで、細胞内のライソゾームに含まれる酵素ARSAをコードする遺伝子の欠損が原因で起こる常染色体潜性遺伝疾患である。白質変性症に対する遺伝子治療においては、すでにex vivoの手法(患者骨髄から取り出し、培養した血球系幹細胞にレンチウィルスを用いてARSA遺伝子を体外で導入し、これを用いて自家骨髄移植治療を行う)が実用化されており、まさに「治せる」疾患の1つとなっているが、まだ適応の制限や限界、あるいは困難が多い。より簡便な治療法としてAAVの髄腔内投与によるin vivo遺伝子治療薬が開発され、治験が行われたが、残念ながら失敗した。その原因として、ヒトMLD患者の症状を改善させるには、従来想定されたレベルより広範囲かつ高いレベルのARSA発現が必要であることが考えられた。
本研究は米国のバイオ企業からの報告で、新たなAAV血清型HSC15を用いることで、この問題を解決することができることが示されている。この血清型は自然界に存在するヒト由来のAAVであり、以前の報告で齧歯類や霊長類において、静脈内投与で血液脳関門(BBB)を効率的に超えて脳内に入り、目的遺伝子を発現させることが可能であることが示されていた(JL Ellsworth et al. 2019)。本報告では、このAAV HSC15を用いてMLDモデルマウスに対する治療効果の検証を脳室内および静脈内投与の2つの経路を用いてAAV9と比較検証した。その結果、HSC15はAAV9に比べてどちらの投与法でも有意なARSAの発現上昇を認め、そのレベルは野生型マウスのそれに迫るものであった。MLDモデルマウスでは運動機能の改善、バイオマーカーの改善を認め、発現されたARSAが組織間で移行することも確認された。さらにARSAの発現は長期間に渡って維持されていた。また霊長類(マカクサル)においても、AAV HSC15に組み込まれたARSAは、静脈内投与により広く脳内に分布し、高いレベルでの発現量を実現できることが示された。
近年、多くの新たな合成血清型がBBBを超えて脳内に遺伝子を導入することが可能であると報告されているが、合成物であるため、臨床応用には安全性の担保などの新たな課題が生まれる。今回、用いられたHSC15は自然由来の血清型である点で安全面でのハードルはやや低いと言える。またAAV9に比較し、静脈内投与で非常に高い有効性が示されており、臨床応用の期待が高まる新たな血清型であると言えよう。
文責:井上 健
https://doi.org/10.1523/JNEUROSCI.1829-22.2023
RNA標的型CRISPR-Cas13dシステムを用いたハンチントン病モデルの治療
An RNA-targeting CRISPR–Cas13d system alleviates disease-related phenotypes in Huntington’s disease models.
K.H. Morelli et al. Nature Neuroscience. 2023, 26; 27–38.
メッセンジャーRNAを標的とした遺伝子発現抑制は、遺伝子治療の1つの手法として研究開発が進み、一部は薬として実用化されつつある。これまでアンチセンスオリゴなどの核酸治療薬、shRNA/miRNAなどをAAVなどのウィルスに組み込んだ遺伝子治療の開発が進んでいるが、今回、新たな手法としてCRISPR-Cas13dを用いた手法が本研究で報告された。従来のCRISPR-Cas9を代表とするゲノム編集技術は、DNAを標的とするものが主体であり、劇的な効果の反面、オフターゲット効果が医療面での応用において危惧されてきた。こと核DNAの切断は、ガン化などのリスクを伴う点が臨床応用における懸念材料とされている。
今回、筆者らはハンチントン病の患者由来iPS細胞から分化誘導した神経細胞やCAGリピート延長を持つモデルマウスに対して、リピートが延長したハンチンチンmRNAを特異的に認識し分解するガイドRNAを設計し、このCRISPR-Cas13dを用いて治療した。患者由来細胞でも、モデル動物でも、リピートが延長した変異体mRNAを特異的に分解し、遺伝子発現プロファイルや表現型が改善することを示した。オフターゲット効果も認めず、高い安全性が示された。
従来のDNAを標的とするCRISPR-Casシステムは、サイズが大きいため、AAVベクターに搭載することが困難であったが、このRNA標的型のCRISPR-Cas13dはAAVに搭載可能なサイズであることから、遺伝子治療との相性が良い。今後、核酸治療薬の対抗軸として、臨床応用にむけた開発が進む可能性がある。
文責:井上 健
https://doi.org/10.1038/s41593-022-01207-1
2つの作用を持つAAV遺伝子治療は進行期カナバン病マウスモデルの病態を改善しうる
Dual-function AAV gene therapy reverses late-stage Canavan disease pathology in mice
Fröhlich D. et al. Front. Mol. Neurosci. 15:1061257.
カナバン病(CD)は、脳内に豊富なアミノ酸NAA(N-acetyl-L-aspartate)を代謝する酵素をコードするASPA遺伝子の変異が原因で起こる白質変性症の1つである。CDに対するASPA遺伝子補充療法は、白質変性症に対する遺伝子治療の先駆的な取り組みとして、すでに臨床研究も実施されているが、必ずしも十分な治療効果が得られていなかった。そこで様々な工夫が凝らされた新たな遺伝子治療の手法が試みられている。今回紹介する論文では、NAA代謝酵素ASPAを補充すると同時に、NAAを合成するために必要な酵素NAT8Lの働きを抑制するというもう1つの治療プロセスを、1つの遺伝子治療用のAAVベクターに合わせ持たせることで、その治療効果を高めようとする取り組みについて紹介されている。著者らは、AAVベクターにNAT8Lに対するshRNA/miRNAとコドンの最適化を行ったASPA cDNAを組み合わせて搭載し、CDモデルマウスであるAKOマウスに対する治療を行った。特筆すべきは治療開始時期を症状が全て出現している生後12週から開始したにもかかわらず、この治療により全ての症状の進行が停止し、さらにすでに出現している多くの症状(NAA蓄積やMRI画像所見を含む)を改善した点である。これは、既にCDの症状が出揃った患者に対しても本治療法が有効である可能性を示唆するものであり、本研究の臨床的意義は高いと考えられる。
文責:井上 健
https://doi.org/10.3389/fnmol.2022.1061257
スプライシング依存的フレームシフトを用いた細胞種特異的な遺伝子発現の制御
Cell-specific regulation of gene expression using splicing-dependent frameshifting
Jonathan P. Ling et al. Nature Communications | (2022)13:5773
AAVを用いた遺伝子治療において、組織あるいは細胞種特異的に必要な遺伝子を発現させることは、治療効果と安全性を高めるために重要であるが、技術的には依然として困難である。これまでの技術として、ウィルスベクターの血清型の指向性や細胞種特異的ミニプロモーターが用いられてきたが、これらのみで厳密な細胞種特異性を発揮することは困難である。本研究は、これらに加えて用いることができる新たな技術の開発に関する報告である。選択的スプライシングは細胞のサブタイプや時期に特異的な遺伝子発現に重要な役割を担っていることが知られている。筆者らは、トランスクリプトミクス・データベースの検索から、細胞種特異的な選択的スプライシングにより包含あるいは除外されるエクソンを選択し、さらにこの可変エクソンを含む、あるいは含まないトランスクリプトにフレームシフトが生ずるように変異を挿入することで、標的細胞のみで特定のフレームの翻訳が起こるようなシステムを構築し、これをsplicing-linked expression design (SLED)と名付けた。SLEDを用いることで、例えばパワフルな広域プロモーターの転写活性を損なうことなく、ニューロンや視細胞での発現の選択性を確保することができる。AAVに組み込んだSLEDベクターは、蛍光タンパク質やカルシウム・インジケーターをニューロンの興奮性/抑制性のサブタイプ別に選択的に発現することもできる。遺伝子治療への応用として、広域プロモーターを用いて視細胞特異的に遺伝子を発現させ、細胞特異的プロモーターを用いた従来型の遺伝子治療と同等の治療効果を発揮したり、腫瘍細胞特異的に細胞死誘導遺伝子を発現させたりすることが可能であることが示された。本法の欠点として、SLEDがイントロンを含むため、DNAサイズが比較的大きいためAAVへの応用が困難になることがある。今後、様々な細胞種のバリエーションとサイズを小さく改変したSLEDが開発されるとより有用性が高まると期待される。
文責:井上 健
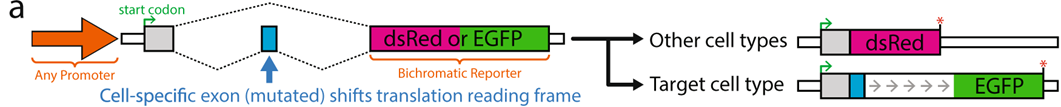
https://doi.org/10.1038/s41467-022-33523-2
脳血管関門を標的とした遺伝子治療によるMCT8欠損症モデルの神経学的症状の改善
Gene therapy targeting the blood–brain barrier improves neurological symptoms in a model of genetic MCT8 deficiency
Sundaram SM et al. Brain 2022 in press.
MCT8欠損症は、Allan-Herndon-Dudley症候群とも呼ばれており、比較的頻度が高い先天性大脳白質形成不全症の1つである。原因遺伝子MCT8は甲状腺ホルモンを細胞内に取り込むため必要なトランスポータータンパク質をコードする遺伝子である。甲状腺ホルモンは、脳においても重要な働きをすることが知られており、MCT8欠損症では、血液の中にある甲状腺ホルモンが脳に入っていかないことが原因であることが知られている。髄鞘化にも甲状腺ホルモンが必須であり、MCT8欠損症では髄鞘化が適切に進まない。これまでにMCT8を介さずに脳の中に入っていくと考えられる甲状腺ホルモンに類似する化合物(トライアックなど)が薬剤候補として開発され、海外では治験が開始されている。これとは別に、アデノ随伴ウィルス(AAV)によりMCT8遺伝子を導入する遺伝子治療法の開発の試みがされているが、どの細胞を標的にして遺伝子治療をするのが最も効果的なのかは明らかではなかった。今回、筆者らは脳の神経細胞やグリア細胞ではなく、脳の血管内皮細胞を標的とすることで、高い治療効果が得られることを報告した。血管内皮細胞は、脳血管関門と呼ばれる血管系と脳を隔絶する障壁の血管側にある細胞で、MCT8が持続的に発現しており、甲状腺ホルモンを血管から脳の中に効率的に通過させる働きをしている。今回、筆者らはMCT8欠損症のモデルマウスに対して、この血管内皮細胞だけを標的とするアデノ随伴ウィルス(AAV)にMCT8を搭載して、静脈内投与により治療を行なった。その結果、モデルマウスでは脳の中に甲状腺ホルモンが効果的に導入され、神経細胞の障害や髄鞘化の障害を改善し、さらに甲状腺ホルモンに依存的な遺伝子の発現や動物の運動障害も改善した。これらの効果の一部は、ある程度症状が出現した後に治療を行なっても再現できた。脳の疾患の遺伝子治療に共通する問題は、AAVをいかに脳血管関門を超えて脳の中の送り届けることができるか、という点であるが、MCT8欠損症については、脳血管関門にある血管内皮細胞を標的とすることで、実に効果的な治療が可能になるということが示された。今後のMCT8欠損症の治療法開発に関する大きな方向性が示された研究成果である。
文責:井上 健
https://doi.org/10.1093/brain/awac243
ナンセンスコドンに対するAAVを用いたsuppressor tRNA遺伝子治療
AAV-delivered suppressor tRNA overcomes a nonsense mutation in mice
J. Wang et al.
Nature 2022 Apr;604(7905):343-348.
AAVによる遺伝子治療の限界として、導入する遺伝子のサイズに制限があること、遺伝子発現量が過剰になること、などの問題点が知られている。この論文では、ナンセンスコドン(読み枠がストップコドンになり、翻訳が止まってしまう変異で、ヒトの疾患遺伝子変異の約11%を占める。)に対して、サプレッサーtRNAをAAVに組み込んで導入することで、安全かつ効果的にナンセンスコドンが原因の疾患モデルマウスを治療できることが報告された。この効果は、単回投与で少なくとも6ヶ月間持続することが示された。
サプレッサーtRNAは、人工的に設計されたtRNAで、ストップコドンに対するアンチコドンを通常のtRNAに組み込んだものである。コンセプトは40年前に報告されたが、治療的有用性は培養細胞での検証にとどまっていた。筆者らは、今回、このサプレッサーtRNAを最適化し、Tyr tRNAを用いたサプレッサーtRNA が最も効率よくストップコドンのリードスルーを誘導することを示した。また同様のリードスルーを誘導するアミノグリコシド系の薬剤と比較して、野生型遺伝子のストップコドンのリードスルーの誘導は低く、オフターゲット効果が低いことが示された。次にこのサプレッサーtRNA をAAVに組み込んで、ムコ多糖症I型の原因遺伝子IDUAにW402X変異を持つモデルマウスに投与し、その治療的有効性を証明した。またNMD標的遺伝子の発現、小胞体ストレスの惹起など起こり得る副反応についての検証も行われ、安全性についての評価も行われた。
サプレッサーtRNAによるAAV遺伝子治療AAV-NoSTOPは、多くの遺伝性疾患の治療の可能性を広げるものである。AAV-NoSTOPは、通常の遺伝子導入治療で見られる過剰発現による毒性(例えばMECP2遺伝子など)の問題を避けることができる選択肢となり得る。また、疾患特異的、遺伝子特異的な治療ではなく、ユニバーサルな治療薬となり得る点も大きなメリットであろう。
文責:井上 健
doi: 10.1038/s41586-022-04533-3.
テイ・サックス病患者に対するAAV遺伝子治療
(AAV gene therapy for Tay-Sachs disease)
Nat Med. 2022 Feb;28(2):251-259.
これまで治療法がなかった遺伝性難治性疾患に対するアデノ随伴ウイルス(AAV)遺伝子治療の実用化が進んでいる。本稿では代表的なライソゾーム病の1つであるテイ・サックス病患者に対するAAVを用いた遺伝子治療のファースト・イン・ヒューマンの結果が報告されている。テイ・サックス病はヘキソサミニダーゼA(HexA)の欠乏によって引き起こされる常染色体劣性遺伝の疾患で、進行性に重篤な神経障害をきたす。本研究では、一次エンドポイントを安全性とし、二次エンドポイントは設定されていない。2名のテイ・サックス病の乳児が対象である。患者1(30ヶ月齢)では、AAVrh8-HEXAおよびAAVrh8-HEXB等モル混合液を髄腔内(大槽と胸腰椎接合部)で総投与量(1×1014ベクターゲノム(vg))が投与された。患者2(7か月齢)では、両側視床(1.5×1012 vg×2)と髄腔内(3.9×1013 vg)に投与された。両患者とも免疫抑制薬が投与された。手技に対する忍容性は高く、ベクター関連の有害事象はなかった。脳脊髄液(CSF)のHexA活性はベースラインから軽度に増加し、安定を維持した。患者2は、髄鞘形成が進行し、注射後3か月までに疾患の安定化を示し、乳児テイ・サックス病の自然歴と比べて一時的な改善を示したが、治療後6か月で疾患の進行が明らかとなった。患者1は、治療前と同じ抗けいれん薬治療を継続し、5歳まで発作はなかった。 患者2は、2歳で抗けいれん薬反応性発作を発症した。著者らは、この研究がテイ・サックス病治療のためのAAV遺伝子治療の早期の安全性と概念実証データを提供したと述べているが、治療よる酵素活性の上昇も画像および臨床症状の改善効果も限定的であったことから、実用化のためには解決すべき技術的な課題があることが明らかとなった。
https://doi.org/10.1038/s41591-021-01664-4
文責:井上 健
アレキサンダー病ラットモデルに対するアンチセンス治療はGFAP病理所見、白質欠損、運動障害を回復させる
(Antisense therapy in a rat model of Alexander disease reverses GFAP pathology, white matter deficits, and motor impairment)
Hagemann et al. Sci. Transl. Med. 13, eabg4711 (2021)
アレキサンダー病(AxD)は、代表的な遺伝性白質変性症の1つである。脳の中にあるグリア系細胞の1つ、アストロサイトに特異的に発現する遺伝子GFAPに変異が起こることで、異常なGFAPタンパク質が過剰に産生され、これがアストロサイトに対して毒性を持つため、大脳白質の変性が生ずる。これまでに有効な治療法がない、難病の1つである。筆者らは、以前、AxDモデルマウスにアンチセンスオリゴ(ASO)を用いてGFAPタンパク質の発現を抑制することで、この疾患に特徴的な脳の病変を軽減することができることを報告した。しかし、このモデルマウスは表現型が軽症で、ヒトのAxD患者に見られるような大脳白質変性や運動機能障害といった症状は見られないため、本治療法が本当にAxD症状を軽減することができるのか不明であった。そこで本研究で筆者らは新たにラットAxDモデルを創出し、ASO治療の有用性について検証を行った。ゲノム編集を用いて、ラットのGFAP遺伝子に重症型AxD患者で見出されたR239H変異を導入した。このラットモデルは、AxD患者脳組織に特徴的に認められる病理学的所見を有する。出生後しばらくは正常であるが、数週間後から運動症状を来たし、14%の動物が2〜3ヶ月で死亡する。このラットモデルにGFAPを標的とするASOを脳室内に1回投与するのみでこれらの症状が軽減することが示された。特記すべきは、ASOによる治療は早期投与によりAxD症状が生ずることを予防するのみならず、すでに症状がある動物に投与した場合でもこれらを回復させることができるという点である。ASOは、脊髄性筋萎縮症やデュシェン型筋ジストロフィーに対する治療薬としてすでに実用化されている。AxDについても国際治験が開始されており、実用化が近いと思われる。
2022.3.4 文責:井上 健
DOI: 10.1126/scitranslmed.abg4711
低分子化合物によってスプライシングを誘導することで制御された遺伝子治療法の開発
(Regulated control of gene therapies by drug-induced splicing)
Nature 2021 Aug 12; 596, 291–295.
これまで、AAVなどを用いた遺伝子治療において、遺伝子の発現レベルを細かく制御することは困難でした。筆者らはこの研究で、ある小分子(薬)を用いた選択的スプライシングの制御が可能な応用度の高いスイッチシステムXon を開発しました。Xonを発現させたい遺伝子の上流に設置することで、AAVからの目的遺伝子の翻訳レベルを薬剤で制御できること報告しています。重要な点は、Xonをオンにするために用いる薬剤LMI070は、ヒトで安全に使用することができ、経口投与が可能で、末梢組織のみならず脳にも分布することです。Xonは、LMI070により選択的スプライシングが変化し、翻訳開始コドンを含む新たなエクソンが挿入されることで、目的遺伝子の発現が可能になるシステムです。遺伝子発現の誘導は、LMI070の単回経口投与で可能であり、発現量は薬物投与量に依存的です。繰り返し投与によって1回目投与と同レベルの発現を繰り返し誘導できます。筆者らは、LMI070単回投与による肝臓でのGFP発現誘導(1900倍)、繰り返し投与によるエリスロポエチン遺伝子治療を介した赤血球増産の長期維持、そして中枢神経系においても神経細胞特異的プロモーターとの組み合わせでプログラニュリン遺伝子の2300倍の発現を誘導することができることを報告しています。AAVからのタンパク質発現の時間的制御を可能にするXonシステムは、細胞生物学や動物を用いた研究への応用が期待されます。さらに使用される薬剤LMI070の経口バイオアベイラビリティと安全性は、Xonスイッチシステムをヒトにおける遺伝子治療に即座に応用することを可能にしますので、今後の遺伝子治療の幅が広がると考えられます。この技術は、例えば適切な発現量の調節が必要な疾患の遺伝子治療法の開発や、一時的にだけ目的タンパク質を発現したい場合(例えばゲノム編集)など様々な分野での応用が期待されます。
2022.1.7 文責:井上 健
doi.org/10.1038/s41586-021-03770-2.
マイクロRNAを用いて導入遺伝子を抑制することで霊長類でのAAVベクターによる後根神経節毒性を軽減する
(MicroRNA-mediated inhibition of transgene expression reduces dorsal root ganglion toxicity by AAV vectors in primates.)
Sci Transl Med. 2020 Nov 11;12(569):eaba9188.
難病の遺伝子治療のためのツールとして期待されるアデノ随伴ウイルス(AAV)ベクターは、他のウィルスベクターに比較して安全性が高いことが知られている。しかし、近年非ヒト霊長類(NHP)の中枢神経系に血液または脳脊髄液を介して送達すると、高い確率で後根神経節(DRG)の毒性を引き起こすことが知られるようになり、ヒトに対するAAV遺伝子治療法の安全性に関する懸念材料の1つとなっている。このDRG毒性は、免疫抑制薬では抑止できないことから、筆者らはこれがDRGニューロンに対する高い形質導入率と過剰な導入遺伝子産物のよる細胞ストレスによって引き起こされるのではないかと考えた(Fig.S2a)。この仮説を検証し、さらにこのDRG毒性を排除するアプローチを開発するために、DRGニューロンに限定して発現しているmicroRNA(miR)183複合体の内因性発現を利用して、DRGニューロン特異的に導入遺伝子発現を抑制した(Fig.S2b)。すなわちmiR183の標的配列をベクターゲノム内の導入遺伝子メッセンジャーRNAの3 '非翻訳領域内に導入した。このベクターをNHPの大槽に注入したところ、未修飾のAAVベクターの投与は、DRGへの強力な形質導入とステロイド抵抗性のDRGニューロンの毒性をもたらした。一方でベクターにmiR183ターゲットを含めると、脳の他の場所での形質導入に影響を与えることなく、DRGニューロンでの導入遺伝子の発現と毒性が低下した。このアプローチにより、AAVによる遺伝子導入がもたらすDRG毒性とこれが関連する罹患の発生率を低減できる可能性があり、多くの中枢神経系疾患に対するより安全性の高いAAV遺伝子治療法の開発を促進するであろう。
文責:井上 健
DOI: 10.1126/scitranslmed.aba9188.
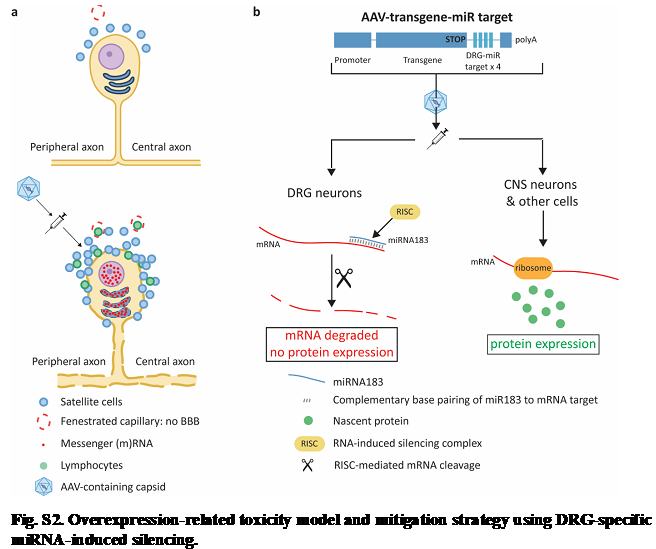
shRNA搭載AAV9による遺伝性ニューロパチーの遺伝子発現抑制治療法の開発
(AAV2/9-mediated silencing of PMP22 prevents the development of pathological features in a rat model of Charcot-Marie-Tooth disease 1 A.)
Nature Communication. 2021 Apr 21;12(1):2356.
Charcot-Marie-Tooth病1A型(CMT1A)は、最も頻度が高い遺伝性疾患の1つで、常染色体優性形式をとる。17番染色体に位置するPMP22遺伝子の重複が主な原因である。これまで有効な治療法は開発されていない。そこで筆者らは、組み替えアデノ随伴ウィルス(AAV)にGFPとPMP22遺伝子を標的としたshRNA(RNA干渉により遺伝子の発現を抑制できる小さなRNA)を組み込み、CMT1Aのモデルラットの坐骨神経への直接投与においてその安全性と有用性の検討を行った。血清型はAAV9が用いられた。投与されたAAVは坐骨神経に分布するシュワン細胞に広がり、過剰発現されたPMP22を正常量に修正し、ミエリンを再生し、電気生理学的所見や運動感覚神経機能を改善させた。オフターゲット効果も免疫反応も限定的であった。さらにヒト患者皮膚から同定されたバイオマーカーが、ラットにおいても治療の有無の判定に有効であった。
文責:井上 健