Prader-Willi症候群(プラダー・ウィリ症候群)
(Prader-Willi Syndrome)
[Synonyms:Prader-Labhart-Willi Syndrome]
Gene Reviews著者: Daniel J Driscoll, MD, PhD, FFACMGG, FAAP, Jennifer L Miller, MD, MS, FAAP, and Suzanne B Cassidy, MD, FFACMGG.
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2023.3.9. 日本語訳最終更新日: 2023.7.30.
要約
疾患の特徴
Prader-Willi症候群(PWS)は、乳児期初期の重度の筋緊張低下、食欲不振、摂食障害に続いて、幼児期には過食のため徐々に病的肥満が生じる(食物摂取を厳密に制限した場合を除く)ことを特徴とする疾患である。運動に関する発達指標や、言語発達は遅延する。一定程度の高次脳機能障害が全例でみられる。性腺機能低下が男女ともにみられ、性器発達不全、思春期発達不全に加え、大多数の例では不妊の形で現れる。低身長が多くみられる(成長ホルモン治療がなされなかった場合)。独特の行動型(癇癪発作,頑固さ,他者を操ろうとする行動,強迫性)が多くみられる。特徴的顔貌、斜視、脊柱側彎もしばしば出現する。
診断・検査
PWSは、15q11.2-q13にあるPrader-Williクリティカル領域(PWCR)内のDNAメチル化異常に起因して生じる一種の隣接遺伝子症候群である。発端者における診断と分子レベルの原因確定は、DNAメチル化解析とオリゴ-SNPコンビネーションアレイ(OSA)を同時施行することで可能である。DNAメチル化解析では、PWCRが母性インプリンティングのみとなっていることが同定される。OSAでは、15q11.2-q13の欠失、インプリンティングセンターの欠失、片親性アイソダイソミー、セグメンタルアイソダイソミーといったものを同定することができる。OSAで異常がみられず、DNAメチル化解析で母性インプリンティングのみであることが判明した例については、DNA多型解析を行うことで、片親性ヘテロダイソミーと、エピ変異によるインプリンティング異常とを鑑別することが可能である。
臨床的マネジメント
症状に対する治療:
乳児期には、十分な栄養状態を確保するため、特殊乳首や経鼻栄養を行う。小児期には、過度の体重増加を抑え(ボディマス指数[BMI]のZ値を2未満に維持し)つつ、エネルギー上の要求も満たせるよう、身長、体重、BMIに基づいた1日の食物摂取量の厳密な管理を行うとともに、運動の奨励を行う。
以下の管理を行う。
- 発達サービスや特別支援教育の活用
- 停留精巣に対するホルモン治療や外科的治療の検討
- 身長の正常化、除脂肪体重の増加と運動性の確保、脂肪量の減少といったことを目的として行う成長ホルモン治療
- 思春期における性ホルモン補充などの内分泌の管理
- 思春期早発症、2型糖尿病、甲状腺機能低下を有する例についてはそれらに関する治療
- 急性の消化器症状を伴う例についてはそれに対する緊急の評価
- 皮膚むしり症に対しては、必要に応じトピラマートもしくはN-アセチルシステインの使用
- 神経行動学的症状と眼科的症状、睡眠の問題、脊柱側彎、股関節形成不全、てんかん発作に対しては標準治療
- 日中の眠気に対するモダフィニルの使用(これが有効な場合あり)
- 骨粗鬆症を避けるためのカルシウムとビタミンDの補充
- 骨密度の低下に対しては性ステロイド、成長ホルモン、ビスホスホネート
- 口腔乾燥症関連製品の使用と口腔衛生の励行
- ソーシャルワーカーの支援とケアコーディネーション
成人期には、PWS罹患者のための生活施設を利用して行動の制御や体重管理を行うことが病的肥満の予防につながる場合がある。また、筋肉量の維持には成長ホルモンが有用な場合がある。
定期的追跡評価:
発達、成長、皮膚、睡眠の問題、家族のニーズ等の状況に関するモニタリングを来院ごとに行う。男性については、精巣の位置を年に1度;思春期、肥満、急激な体重増加がみられる例については、糖化ヘモグロビンの評価やブドウ糖負荷試験;遊離T4とTSHの評価を6-12か月に1度行う。中枢性副腎機能不全の評価を必要に応じて;身長、体重、BMIの評価を乳児期は月に1度、10歳までは6ヵ月に1度、その後は年に1度行う。行動の問題に関する評価を2歳以降、年に1度、精神病に関する評価を思春期から成人期にかけて年に1度行う。視力ならびに睡眠の問題に関する評価を年に1度;睡眠検査を、成長ホルモン治療開始前と開始後4-8週後に行う。脊柱側彎に関する臨床的診査を、子どもがひとり座りができるようになったら、来院ごとに;脊柱側彎の臨床所見あるいは肥満を有する例については、脊椎のX線写真を年に1度;DEXAスキャンを、思春期から始め、2年に1度行う。新たなてんかん発作の有無に関する評価、もともと発作を有していた例については状況の変化に関する評価を来院ごとに行う。歯科的問題を抱える例については、歯科的評価を6ヵ月ごと、もしくはこれよりさらに高頻度に行う。
遺伝カウンセリング
PWS罹患者は、通常、孤発例(家系内で1例だけの発生)で、denovoの遺伝学的変化の結果として本疾患に至ったものである。大多数の家系については、再発リスクは1%未満であるが、病因によっては再発リスクが50%に達するものもあり、また、きわめて稀ながら、リスクが100%に近いシナリオも理論上は描きうる。こうしたことから、PWSの再発リスク評価を信頼性のあるものにする上では、発端者におけるPWSの遺伝学的メカニズム(すなわち、15qの欠失,UPD15,インプリンティング障害)の特定、ならびに、素因となりうる遺伝学的変化(例えば、片親の染色体再構成、あるいは父性のヘテロ接合性インプリンティングセンター欠失)を明らかにするための両親に対する検査が必要となる。発端者において原因となった遺伝学的メカニズムが特定されれば、PWSに関する出生前検査が可能となる。
診断
本疾患を示唆する所見
以下のような特徴的な臨床所見や臨床検査所見を有する例については、Prader-Willi症候群を疑う必要がある。
臨床所見
臨床所見は年齢ごとに異なってくる。その年齢に該当する例が、その年齢でみられる所見を呈する場合は、PWSに関する分子レベルの解析を行うべきであると考えられる(「診断の確定」の項を参照)。
新生児期
吸啜力の低下を伴う筋緊張低下
生後1ヵ月-2歳未満
- 新生児期の食欲不振と哺乳障害を伴う筋緊張低下
- 発達遅滞
2歳―6歳
- 吸啜障害の既往を伴う筋緊張低下
- 発達遅滞
6歳―12歳
- 吸啜障害と筋緊張低下の既往(筋緊張低下は長く続くことがある)
- 発達遅滞
- 中枢性肥満を伴う過食(外からのコントロールがなされていない場合)
13歳―成人期
- 高次脳機能障害(知的障害の程度は通常、軽度)
- 食餌量の増加と中枢性の過食症(外からのコントロールがなされていない場合)
- 視床下部性性腺機能低下症、ないし特徴的行動所見
検査所見
15q11.2-q13領域の欠失は、PWSを示唆するものではあるが、PWSの診断を確定させるものではない。
診断の確定
発端者におけるPWSの診断は、15q11.2-q13にあるPrader-Williクリティカル領域(PWCR)内に、以下に挙げるものの1つに起因して生じた、母性インプリンティングのみというDNAメチル化異常が同定されることをもって確定する。
- 父性継承の15q11.2-q13領域の欠失
- 15q11.2-q13領域の母性片親性ダイソミー(UPD15)
- インプリンティングセンターの欠失、エピ変異のいずれかに起因して生じた父性継承の15q11.2-q13領域のインプリンティング異常
分子遺伝学的検査
分子遺伝学的検査には、第1段階の検査、第2段階の検査、その他の検査がある。
推奨される第1段階の検査
検査は、まず、DNAメチル化解析と、オリゴ-少塩基多型(SNP)コンビネーションアレイ(OSA)を同時施行する。これにより、大多数の例で診断の確定や分子レベルの原因確定が可能である(図1ならびに表1参照)。
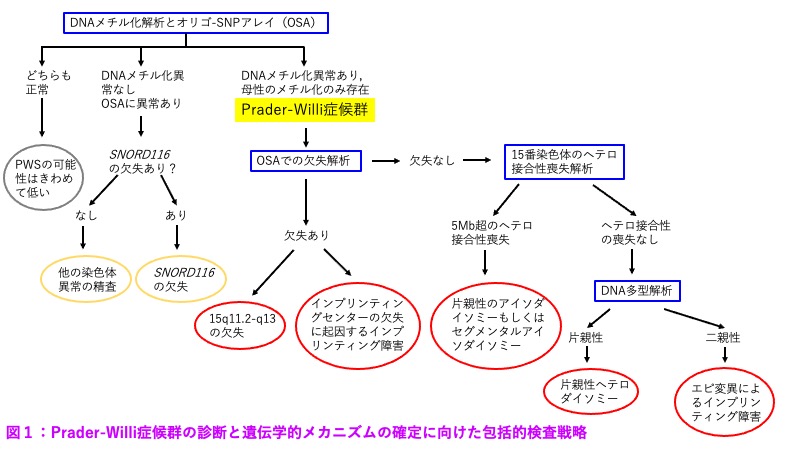
図1:Prader-Willi症候群の診断と遺伝的メカニズムの確定に向けた包括的検査戦略
AOH=ヘテロ接合の欠如;chromabn=染色体異常;IC=インプリンティングセンター;OSA=オリゴ-SNPアレイ;PWS=Prader-Willi症候群;UGD=片親性ダイソミー;W/U=精密検査
- DNAメチル化解析
これは通常、メチル化特異的PCR(MSP)で行われる。これを行って、15q11.2-q13が母性インプリンティングのみになっていることを同定することで、PWSの診断を確定させるところまでは可能であるものの、DNAメチル化異常の原因を特定することまではできない(15qの欠失、UPD15、インプリンティング異常のそれぞれを区別することまではできない)。
- OSA
OSAというのは、オリゴヌクレオチドとSNPのプローブを組み合わせて使用して欠失/重複を検出し、1型の欠失と2型の欠失、さらには非定型的欠失も含めた鑑別を行う(図2参照)とともに、その他に何らかの重要な染色体異常(不均衡型転座という稀な例でみられる異常)がないかを調べようというものである。現在用いられているOSAの多くは、小さなインプリンティングセンターの欠失や、PWSと一部の症候が重なる他の遺伝性疾患(例えば、SNORD116遺伝子クラスターの欠失;「鑑別診断」の項を参照)を同時に検出する能力を有する。さらに言うと、OSAは、アイソダイソミーやセグメンタルアイソダイソミーに起因して生じたUPD15の例をも同定しうる能力を有する。
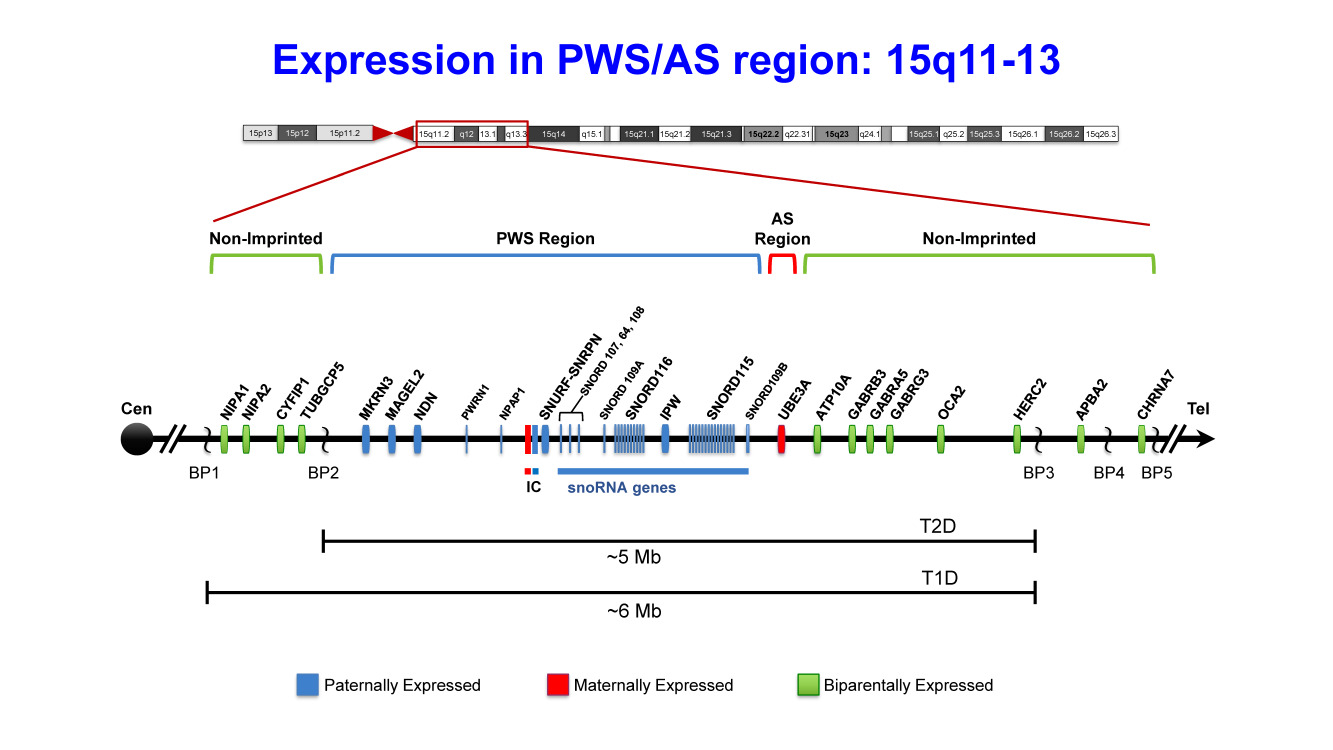
図2:15q11.2-q13.3領域の遺伝子マップ,発現マップの概要
Prader-Willi症候群(PWS)クリティカル領域(青色で示した)には、いくつかの父性発現遺伝子(MKRN3,MAGEL2,NDN,PWRN1,NPAP1,SNURF-SNRPN)、父性発現の核小体低分子RNA(snoRNA)遺伝子ファミリー、長鎖ノンコーディングRNAであるIPWがある。これらの遺伝子のうちの3つ(SNURF-SNRPN,MAGEL2,NDN)は、それぞれのプロモーター領域内にメチル化可変CpGアイランドを有しており、これらの母性アレルはメチル化されて抑制状態にある。Angelman症候群(AS)に関連するUBE3A(赤色で示した)だけは母性発現であるが、インプリンティングのなされた発現は、一定の組織特異的領域(特異的に脳)に限定した形でみられる。インプリンティングセンター(IC)は、AS(母性;赤色で示した)とPWS(父性;青色で示した)の2部構成になっている。PWSの最小共通欠失領域(PWS-SRO)は、4.3kbの領域に局在し、その内容は、バイシストロン性遺伝子であるSNURF-SNRPNのプロモーター、CpGアイランド、エクソン1、ならびにイントロン1のごく一部にまたがることがわかっている。AS-SROは、SNURF-SNRPNのエクソン1より35kb近位に存在する。2部構成のインプリンティングセンターは、2.5MbのPWS/ASインプリンティング領域内に存在する。GABA受容体遺伝子群(GABRB3,GABRA5,GABRG3)、ATP10A、OCA2(この遺伝子の病的バリアントにより眼皮膚白皮症2型が生じる)、HERC2はインプリンティングを受けず、二親性の発現を示す(緑色で示した)。横線に縦に入れた区切りは、PWSとASで生じる5Mbと6Mbのありふれた欠失(commondeletion)の3つの切断点、BP1、BP2、BP3を表している。稀には、より遠位にBP4、BP5という切断点が現れることもある。BP1とBP2の中間部には、NIPA1、NIPA2、CYFIP1、TUBGCP5というインプリンティングを受けないさらに別の4つの遺伝子が存在する。PWS罹患者でみられる欠失の90%超を、1型と2型の欠失が占める。1型の欠失(T1D)はBP1からBP3まで、2型の欠失(T2D)はBP2からBP3までの欠失である。SNORD116とSNORD115には、ここに示したより数多くの、それぞれおおむね24コピー、47コピーが存在する。すなわち、このマップは実際の縮尺に忠実に描いたものではないことに注意されたい。このマップの並びは、最新のヒトゲノムアセンブリ(UCSCGenomeBrowser,GRCh38/hg38)に基づくものである。
推奨される第2段階の検査
15q11.2-q13が母性インプリンティングのみになるというDNAメチル化異常を有するものの、OSAで15qの欠失やUPD15を示さない(ヘテロ接合性が失われてアイソダイソミーあるいはセグメンタルアイソダイソミーの状態になっているわけではない)例については、ヘテロダイソミーの形で生じたUPD15、あるいはエピジェネティックなインプリンティング異常に起因して生じたDNAメチル化異常を調べるため、発端者とその両親に対してDNA多型解析を行うことが推奨される。
それ以外の検査
- メチル化特異的MLPA法(MS-MLPA)
MS-MLPAは、15q11.2-q13が母性インプリンティングのみになっていることを確認することで、PWSの診断を確定させることができる手法で、同時に、以下の能力も有する。
(1)父性継承の15q11.2-q13領域の欠失の有無とその大まかなサイズを特定できる。
(2)1型欠失と2型欠失を同定できる(図2参照)。
(3)MSPが1領域のみを対象とするのに対し、6つのメチル化可変領域を同時に調べることができる。
(4)しばしば、モザイクの同定やモザイク率の定量が可能。
(5)インプリンティングセンターとSNORD116の欠失を同定できる(図1ならびに「鑑別診断」の項を参照)。
なお、MS-MLPAでは、UPD15とエピ変異によるインプリンティング異常とを区別することはできない。
- DNA配列解析
15q11.2-q13のDNAメチル化異常を有し、母性インプリンティングのみになっていることが判明している例については、DNA配列解析を行うことで、インプリンティングセンターの欠失に起因するインプリンティング異常の同定が可能である。
注:これが実際に行われることはそれほど多くない。
それは、MS-MLPAや大多数のOSAでは、高濃度のプローブを用いることでインプリンティングセンターの欠失の検出が可能だからである。
- FISH解析
FISH法は、使用する特定のプローブ(例えば、SNRPN)に限定した形で行われ、これにより15q11.2-q13の欠失を同定することが可能である。
FISHでは、以下のようなことはできない。
(1)PWCR全体の調査
(2)欠失のサイズの同定
(3)染色体の残りの部分に関する情報取得
(4)UPD15やインプリンティング障害の同定
第1段階の検査としてFISH解析が推奨されるようなことはない。しかし、再発リスクを明確にするという目的では用いられる場合がある(表9ならびに表10を参照)。
注:遺伝カウンセリングの上では、PWSの背景にある遺伝学的病因の違いの見極めが重要である(表9ならびに表10を参照)。
表1:Prader-Willi症候群で用いられる分子遺伝学的検査
| メカニズム1 | このメカニズムに起因するPWSの割合 | DNAメチル化解析2 | OSA3 | DNA多型解析4 | MS-MLPA5 |
|---|---|---|---|---|---|
| 15qの欠失 | 60%-70%近く | DNAメチル化異常あり;具体的な原因の特定は不能 | + | + | |
| UPD15 染色体全体のアイソダイソミー |
4%-5% | + | DNAメチル化異常あり;具体的な原因の特定は不能 | ||
| UPD15 セグメンタルアイソダイソミー |
17%-23% | + | |||
| UPD15 ヘテロダイソミー6 |
8%-11%近く | + | |||
| インプリンティングセンターの欠失 | 0.5%未満 | + | + | ||
| エピ変異によるインプリンティング異常 | 2%-4%近く | + | DNAメチル化異常あり;具体的な原因の特定は不能 |
- 詳細については、「分子遺伝学」の項を参照。
- 通常、メチル化特異的PCR(MSP)で行われる。
- OSAは、配列解析では検出できないような大きな欠失/重複をゲノムワイドに検出することができ、たいていのインプリンティングセンター欠失(図1参照)を含め、欠失のサイズに関する詳細な情報をこれにより知ることができる。さらに、少塩基多型(SNP)のプローブを用いることで、1つの染色体全体のアイソダイソミーやセグメンタルアイソダイソミーの形でのUPD15は検出されるが、ヘテロダイソミーの検出はできない。
- 第1段階の検査でこれを用いるのではなく、DNAメチル化解析によりPWSの診断が確定した後の段階で、UPD15ヘテロダイソミーとエピ変異によるインプリンティング障害とを鑑別する目的で、これが行われる。
- MS-MLPAでは、15qの欠失とたいていのインプリンティングセンターの欠失が検出可能である(図1参照)。 これによりDNAメチル化異常の検出が可能であるが、UPDとエピ変異によるインプリンティング障害との鑑別は、MS-MLPAではできない。
- これは、アイソダイソミーとなる(すなわち、ヘテロ接合性を示さない)領域を一切含まないUPD15をいう。
臨床的特徴
臨床像
Prader-Willi症候群(PWS)は、新生児期における吸啜障害ならびに栄養支援なしでは体重増加不良をきたすような状況を伴う筋緊張低下、発達遅滞、軽度の高次脳機能障害、性器低形成と思春期発達の不全をきたすような性腺機能低下、成長ホルモン(GH)治療が行われなかった場合の低身長、過食の制限が行われなかった場合の小児期に始まる肥満、行動の問題、そして多くの場合、特徴的顔貌が現れる、複雑で多系統に影響が及ぶ疾患である。常にではないものの、比較的多くみられる症候としては、胎動減少、小さな手足、罹患者家族と比べたときの低色素沈着、皮膚むしり症、斜視、視力の異常、睡眠障害(日中の眠気や、時に睡眠時無呼吸)、濃く粘性の高い唾液、構音の変化などがある。PWSの分子レベルの原因の違いによって、臨床症候には多少の幅がみられる。
表2:Prader-Willi症候群:代表的症候の出現頻度
周産期の所見
10歳未満のPWS罹患者47人の出生前超音波を用いた後ろ向き研究にて、罹患者は対照群に比して、胎動が少なく(88%)、在胎不当過小(65%)、頭囲-腹囲比の上昇を伴う不均衡な子宮内発育(43%)、羊水過多(34%)を示した[Grossら2015]。出生前の筋緊張低下は、通常、胎動減少、分娩時の異常胎位、補助分娩・帝王切開の比率の
上昇となって現れる。胎児の大きさは、通常、正常範囲内であるが、出生時の体重とBMIは、ごくふつうの発育状態だった同胞と比べ、平均で15%低い値を示す[Millerら2011]。
筋緊張低下
| 症候 | その症候を有する罹患者の割合 | コメント |
|---|---|---|
| 乳児期の筋緊張低下 | 95%-100% | 摂食支援がない場合は、吸啜障害に起因して体重増加不良をきたす。 |
| 嚥下障害 | 90%-100% | 出生時からみられ、成人期まで続く。 |
| 運動発達の遅延 | 90%-100% | |
| 言語発達の遅延 | 90%-100% | 構音の異常や発語失行など。 |
| 知的障害 | 90%-100% | 大多数は軽度の障害であるものの、重度の学習障害から相当程度の高次脳機能障害まで幅がみられる。 |
| 内分泌症候 | 90%-100% | 性腺機能低下,思春期発達の異常,発育障害,糖尿病,甲状腺機能低下 |
| 過食と肥満 | 90%-100% | |
| 特徴的行動プロフィール | 70%-90% | 不安,癇癪,頑固さ,強迫性障害,他者を操ろうとする行動,自閉症的特徴,注意欠如/多動性障害;精神病は、特にUPD15を有する例で成人初期に現れる。 |
| 疼痛閾値の上昇 | 60%-80% | これにより、緊急の加療を要する事態が表に現れないことがある。 |
| 顔面の形態異常 | 50%-70% | 成長ホルモン治療により軽減していくことがある;15q欠失例でより多くみられる。 |
| 低色素沈着 | 50%-70% | 主として15q欠失例でみられる。 |
| 皮膚むしり症 | 50%-60% | 年長の成人では少なくなる。 |
| 斜視 | 40%-60% | |
| 睡眠の異常 | 30%-40% | 中枢性無呼吸(乳児期),閉塞性睡眠時無呼吸,日中の眠気,ナルコレプシー |
| 脊柱側彎 | 40%-80% | |
| てんかん発作 | 10%-20% | 通常は全般発作で、治療可能。 |
乳児期の筋緊張低下は、ほぼ全例でみられ、その結果、自発覚醒の低下、弱い泣き声、吸啜反射を含めた反射の低下などを伴う体動減少と傾眠が生じる。筋緊張低下の起源は中枢性で、診断目的で筋生検を含めた神経筋の検査を行ったとしても、通常は、異常なし、あるいは廃用性の非特異的変化がみられるのみという結果に終わる。
吸啜障害、嚥下障害、傾眠、食欲不振のため、摂食補助を行わなかった場合、乳児期初期には体重増加不良に陥る。摂食障害が乳児の99%で報告されており、多くの場合、経鼻栄養(稀ながら、胃瘻造設が必要になる場合もある)もしくは特殊乳首の使用が必要になる。
そうしたものが必要になる期間はさまざまであるが、多くは数週から数ヵ月といったところである。子どもがコップで飲んだり、固形物を食べたりする頃までには、ほぼ正常な食生活が送れる時期が訪れる。
筋緊張低下は、経時的に改善していく。ただ、小児期や成人期に至っても、筋肉量や筋緊張度の低下を伴う軽度の筋緊張低下状態は続くことになる。
発達遅滞
初期の運動発達指標の達成は、通常の約2倍の月齢にまでずれ込む(おすわりは12ヵ月、歩行開始は24ヵ月)。言語発達の指標も遅延することが多く、スピーチに障害がみられる[Dimitropoulosら2013]。PWS罹患者の多くで構音の異常がみられる。発語失行が7%-10%で報告されており、15qの欠失を伴う例でこれがより高頻度にみられる。言語発達の極度の遅れを示す例がごく一部にみられるものの、大多数の例について言うと、言語能力はむしろ、相対的に強みをもつ分野である。
知的障害
知的障害は、通常、子どもが就学前期に達するまでに明らかになる。検査を行うと、多くのPWS罹患者は軽度の知的障害を示す(平均IQ:60-70)一方、境界域の知的障害あるいは正常低値の知能を示す例が約40%、中等度の知的障害を有する例が約20%みられる。IQの値とは無関係に、多くのPWS罹患児は複数の重度の学習障害を有し、本人の有している知能以上に、学業成績のほうは芳しくない[Whittington&Holland2017]。著者らの経験した範囲で言うと、PWS罹患者で大学に進んで卒業にまでこぎつけられる割合は、ごくわずかである。特に15qの欠失を有する例については、ジグソーパズルの能力が高いことが報告されている。
内分泌症候
- 性腺機能低下症
男女ともに性腺機能低下がみられ、具体的には、大多数が、性器低形成、思春期発達不全、不妊の形で現れる。性器低形成は、出生時からみられ、生涯続く。
- 男性
- 女性
- 発育不全
性器低形成は見逃されることが多いものの、出生時から、大陰唇・小陰唇・陰核の矮小化が多くみられる。
性腺機能低下は、通常、血清ゴナドトロピン濃度の低下に起因するもので、思春期発達の不全、遅延、さらに時には障害にさえつながることがある。ほぼ全例が不妊であるが、女性については生殖に至った例が少数報告されている[Cassidyら2012]。PWSでみられる性腺機能低下は、長い間、完全に下垂体性のものと考えられてきたが、その後の研究で、下垂体と原発性の性腺の問題とが組み合わさった形で関与していることが示唆されている[Eldar-Gevaら2009,Hirschら2009,Eldar-Gevaら2010,Gross-Tsurら2012]。そうした考え方がなされるようになった主たる背景は、両性とも、一部の罹患者については、ゴナドトロピン分泌低下がみられず、かつインヒビンBが異常な低値を示すという点である。
停留精巣を有するPWS罹患男性乳児については、ヒト絨毛性ゴナドトロピン(hCG)での治療が可能である。これを行うことで、泌尿器の手術を行う以前に、精巣の解剖学的位置を下降させるとともに、陰茎・陰嚢のサイズを改善させることができる。精巣固定術を若年で行うほど、また、hCG治療後のインヒビンB値やテストステロン値が高いほど、精巣の組織学的検査にて、精細管中の生殖細胞数が増加することがわかっている[Bakkerら2015]。
陰毛の早発が男性の15%-20%、女性の30%にみられると報告されている。また、副腎皮質性思春期徴候の早発がPWS罹患者の15%-20%で報告されている。陰毛の早発と副腎皮質性思春期徴候の早発は、ともに、硫酸デヒドロエピアンドロステロン値の上昇に起因して生じることがわかっている。そしてまた、骨年齢の亢進も報告されている。中枢性思春期早発症が罹患者の5%で報告されているが、これは、通常、特発性である。
300人超の罹患児が対象となった15以上の研究データから、PWS罹患者では成長ホルモンの分泌低下が生じるとされる[Burmanら2001]。低身長が、未治療罹患者の60%-70%で報告されている。小児期にはそれほど目立たなかったとしても、成長ホルモン治療なしの場合、10歳代ではほぼ全例で低身長が明らかとなる。思春期性の成長スパートが欠如することになるため、未治療の場合の平均身長は、男性で155㎝、女性で148㎝となる。PWSにおいては、成人でも成長ホルモン分泌低下がみられる[Grugniら2006,Höybye2007]。成長ホルモンの充足の如何にかかわらず、PWS罹患者については成長ホルモン治療を行うことが有益である[Alves&Franco2020,Höybyeら2021]。成長ホルモン治療を受けていない乳児・小児の成長チャートが公表されており[Butlerら2011,Butlerら2015]、成長ホルモン治療を受けたPWS罹患児の成長チャートも作成されている[Butlerら2016]。
手足の成長は遅延し、成長ホルモン治療なしの場合、通常、70%-90%の罹患者については、10歳の段階で5パーセンタイルを下回る。成人期の足のサイズは、平均で、女性が20.3㎝、男性が22.3㎝である。
- 視床下部性下垂体機能不全
視床下部の発生や機能に問題が生じると、成長ホルモン分泌不全、性腺機能低下、甲状腺機能低下、副腎皮質刺激ホルモン分泌不全、オキシトシンニューロンの異常など、複数のホルモン分泌不全をきたす結果となる[Tauber&Hoybye2021]。
- Ⅱ型糖尿病
PWS成人の25%近く(特に顕著な肥満を有する例)がⅡ型糖尿病を有し[Tauber&Hoybye2021]、平均発症年齢は20歳である。早期診断、両親に対する教育、成長ホルモン治療、PWS成人に特化したグループホームの利用等が可能になったことで、病的肥満とⅡ型糖尿病の発生は減少してきている。 - 中枢性甲状腺機能低下
甲状腺刺激ホルモンは正常で、かつ遊離サイロキシンが低値となる中枢性甲状腺機能低下が、PWS罹患者の25%近くで報告されている。診断時の平均年齢は2歳である[Millerら2008,Dieneら2010]。 - 中枢性副腎機能不全(CAI) 当初のオランダの研究で、PWS罹患者にはCAIが多くみられると言われていた[deLindvanWijngaardenら2008]が、その後に行われた複数の研究(そのうちの1つは大規模な国際研究)で、PWSの成人について、CAIは稀である(1.2%)ことが判明した[Rosenbergら2020]。その大規模国際研究は、副腎機能不全の症候がなく、確認もできない場合、PWS罹患者へのヒドロコルチゾンの補充は不要であると結論づけている。
- 食欲,肥満,消化器症候
長い間、PWS罹患者ではっきりと区別しうる栄養学的な相は2つだけ(すなわち、体重増加不良を示す相と、その後の過食・肥満の相)であるとされてきたが、多施設共同研究の結果、2つの相の間の移行段階はこれよりはるかに複雑で、PWS罹患者は通常、7つもの栄養学的な相を経ていくことが判明した[Millerら2011](表3参照)。
表3:Prader-Willi症候群でみられる栄養学的な相
| 相 | 年齢の中央値 | 臨床的特徴 |
|---|---|---|
| 0 | 出生前から出生時 | 胎動の減少と、同胞に比べ低い出生時体重。 |
| 1a | 出生時から9ヵ月 | 摂食障害と食欲不振を伴う筋緊張低下。 |
| 1b | 9ヵ月から25ヵ月 | 摂食と食欲が改善;適正な成長。 |
| 2a | 2.1歳から4.5歳 | 食欲増進や栄養摂取量の増加を伴わない体重増加。 |
| 2b | 4.5歳から8歳 | 食欲と栄養摂取量の増加がみられるものの、満腹感もある。 |
| 3 | 8歳から成人期 | 過食で、満腹感がほとんどない。 |
| 4 | 成人期 | 一部の罹患者では、飽くことを知らない食欲はみられなくなる。 |
Millerら[2011]より引用。
- 過食
PWSでみられる過食は、視床下部の異常が原因で、これにより満腹感が得られなくなるものと考えられている。食物のため込みやあさり回り、食物でないものを食べる、食物や食物購入のための金の窃盗といった食物探索行動が多くみられる。現時点では、過食を引き起こす原因として全例でみられるホルモン異常は同定されておらず、PWSでみられる過食と代謝面で相関をもつ因子は明らかになっていない。
- 肥満
肥満は、過食行動、ならびにカロリー必要量の低下の2つが原因となって生じる。カロリー必要量の低下は、非罹患者と比べ、活動量や除脂肪体重(主として筋肉)が少ないことに起因して、安静時代謝量が少なくなっていることによるものである。PWS罹患者でみられる肥満は、男女とも主として中心性(腹部,臀部,大腿部)である。興味深いことに、肥満者における内臓脂肪の量は、肥満の程度から推測される量より少ない。肥満とその合併症は、罹病や死亡の主要原因となっている(本セクションの最後尾にある「寿命」の項を参照)。
早期診断がなされることで、臨床医は、栄養学的な相(表3参照)を中心に、PWSの自然経過に関する予備的ガイダンスを開始することが可能となり、また家族に対し、肥満のリスクがあること、ならびに体重増加を監視しながら、おおむね18ヵ月から36ヵ月にはカロリー摂取を制限すべきことを伝えることも可能となる。「症候に対する治療」の項で述べている食餌、運動、監視プログラムが実施されれば、肥満は予防できる可能性がある。
良好な食餌コントロールとともに成長ホルモン治療を幼少期に開始することができれば、肥満と脂肪量過多は予防できる可能性がある。また同時に、これにより、PWSに特徴的な顔貌の修正、運動発達指標の改善、一部の認知機能の改善にもつながる可能性がある[Butlerら2019b,Ayet-Rogerら2022]。
- 急性の消化器症候
大多数の罹患者は、胃内容の排出が遅延し(罹患者の60%-80%)、かつ嘔吐は稀である。嘔吐の減少は80%-90%で報告されており、顕著な過食に伴い胃壊死のリスクが高まる。生命を脅かすような胃の炎症や壊死の徴候としては、腹部の膨満、鼓脹、疼痛、不活動、食欲喪失、嘔吐などが考えられる。こうした徴候を呈する例については、医学的評価を迅速に行う必要があり、状況によっては入院が必要になる。X線撮影や、場合によっては緊急手術が必要になるようなこともある。止瀉薬を使用すると、重篤な腸の膨満、壊死、破裂を引き起こすおそれがある。
行動
PWS罹患者においては、不安、癇癪(感情の爆発)、固執/変化に対する抵抗、強迫性行動、社会的認知の障害などの特徴的行動プロフィールが、幼児期には明らかにみられるようになる[Ishiiら2017,Schwartzら2021]。こうした行動は、小児期ならびに思春期に、年齢が上がるにつれ、またBMIが上昇するにつれ顕著になっていくが、Dykens[2013]によると、外に向かう行動上の問題(例えば、攻撃性、衝動性)は、年長の成人(40歳超)になるに従って減少していく傾向にあるという。
- 行動特性の多くは、自閉症を示唆するものとなっている。
PWS罹患者786人を対象として行われた最近のメタアナリシスによると、全体の26.7%(15q欠失例の18.5%,UPD15例の35.3%)が自閉症スペクトラム障害の基準を満たしていたという[Bennettら2015]。
- 注意欠如/多動性障害が多くみられ、早発性である。
- 一部の罹患者では、成人初期に精神病が明らかとなるが、これはUPD15を有する罹患者に有意性をもって高頻度にみられる。5研究、計95人のPWS罹患者のメタアナリシスによると、全体としての精神病の発症率は、15q欠失例で25%、UPD15例で64%であったという[Yangら2013]。
行動の問題や精神医学的問題は、独立した生活を営む能力への悪影響という意味も含め、思春期ならびに成人期の生活の質に関する最も大きな障害となっている。
無痛症
痛み感覚の鈍麻が多くみられ、これにより感染、外傷、骨折といったものが見過ごされてしまう可能性がある。PWS罹患者、状況がよほど悪化しない限り痛みを訴えないことがあることに加え、痛みの部位の特定も難しいことがある。
形態異常
特徴的顔貌(前頭部幅径の狭小化,アーモンド形の眼瞼裂,狭い鼻梁,口角の下降を伴う薄い上赤唇)は、出生時に明らかにみられる場合もあれば、そうでない場合もある。これらの特徴は、時間の経過とともに徐々に顕著になっていく[Cassidy&Driscoll2009,Cassidyら2012]。成長ホルモン治療を行うと、そうした特徴は経時的に薄らいでいくことがある。
15q欠失例については、15qに座位するOCA2の欠失に起因して、毛髪・眼・皮膚の低色素沈着がしばしばみられる。
皮膚症候
皮膚や粘膜に対するむしり症が、しばしば慢性の開放創となって残り、これが厄介な問題の1つになることがある。鼻、直腸、膣のむしり症が多くみられ、しばしば介護者に気づかれることなく経過することになる。むしり症により、色素性変化、瘢痕形成、感染症といったものが引き起こされることがある。
肥満のPWS罹患者については末梢性浮腫が珍しくなく、下肢の慢性変化につながることがある。
眼科的症候
PWS罹患者にみられる斜視は、5歳までに診断がなされることが多い。近視や遠視も多くみられる[Bohonowychら2021]。
睡眠の異常
REM潜時の減少、睡眠構造の変化、酸素飽和度の低下、中枢性・閉塞性両面の無呼吸をはじめとする睡眠の異常がよく知られている[Festenら2006,Prianoら2006]。睡眠微細構造の変化や睡眠中の換気異常を引き起こしているのは、原発性視床下部機能障害であると考えられている。PWS罹患者の中には、短時間でのREM睡眠への移行とnon-REM睡眠の不安定性の低下を伴って、ナルコレプシー類似の日中の過度の眠気を示す例がみられる[Bruniら2010]。ナルコレプシーは、10%-35%にみられるとされているが、日中の傾眠の多さから考えて、PWS罹患者においてはナルコレプシーが診断されないままになっていることが多いように思われる。ナルコレプシーの診断には、複数の睡眠潜時の検査が不可欠である。PWS罹患者ではカタプレキシーが生じることが知られているが、その発生頻度はわかっていない。
骨格所見
脊柱側彎は、罹患者の40%-80%に認められるが、その発症年齢と重症度には幅があり、中には乳児期にすでに存在するような例もみられる。背景に特段の構造奇形が存在しないことから、これは筋緊張低下に起因するものと考えられる。思春期ならびに成人のPWS罹患者には脊柱後彎も多くみられる。股関節形成不全が、おおむね20%-30%にみられる[Triznoら2018]。PWSでは骨減少症や骨粗鬆症の増加もみられる。骨折頻度、特に長管骨の骨折頻度に上昇がみられるようにも思われるが、今のところ、これに関し、研究デザインの整った報告はみられない。
てんかん発作
てんかん発作(通常は全般発作)が幼児期に生じることがある。頻回に生じる例は少なく、抗痙攣薬を数年間使用することで治癒することが多い[Takeshitaら2013]。
歯科的問題
PWS罹患者では、対照群に比べ唾液流出が減少し、齲蝕の増加や構音障害につながることがある。口唇の乾燥組織形成が多くみられる。歯の叢生やエナメル質形成不全も、一般より多く見受けられる。
寿命
PWS罹患者の死亡率は、知的障害を有する対照群と比べ、高い値を示す。ただ、治療の進歩により、死亡率は年あたり1.25%にまで減少してきている[Whittingtonら2015]。いくつかの大規模研究から、小児の死亡については呼吸不全その他の熱性疾患、成人については心疾患・心不全、肺血栓塞栓、肥満関連の合併症、胃の問題が、死亡原因として最も多く関与していることがわかっている[Butlerら2017,PacoriconaAlfasoら2019]。注目すべきものとして、Prader-Willi症候群協会(アメリカ)の把握している2,000人超(うち114人はすでに死亡)の罹患者の家族に対してアンケートの形で行った調査がある。その内容は、死亡者のほうが生存者より年齢が高いことを差し引いてデータの調整を行ったとしても、なお生存者は死亡者に比べ成長ホルモン治療施行率が高い(3倍)というものであった[Proffittら2019]。
PWS罹患者、特に、今は痩せているが以前は肥満であった人が暴食を行ったというような例で、胃の膨満により胃破裂や胃壊死に至ったとする報告がみられる。ただ、そうしたことも、疼痛閾値が高いため気づかれないままになることがある。
窒息、特にホットドッグによる窒息が、PWS罹患者の死亡の約8%を占めるとの報告がみられる。PWSにおいては、嚥下の咽頭期ならびに食道期に異常が多くみられ、また、残存物を排出しようとしたり咳とともに出したりといった行動が欠如することも多い[Grossら2017]。こうしたことと早食いとがあいまって、誤嚥関連の死亡リスクを高めていると考えられる。
成長ホルモン治療を開始して数ヵ月のうちに死亡した例が複数報告されており、成長ホルモン投与によって不測死が生じているのではないかとの懸念が提起されている。
数少ない死亡例の大半は、もともと呼吸器疾患あるいは心疾患を有していた上に、上気道閉塞があり、かつ扁桃・アデノイド肥大に対する処置がなされていなかった肥満者である。
ただ、成長ホルモン治療の有無で罹患者の死亡率に違いはみられなかったとする報告も複数存在する。こうしたことから、成長ホルモン治療投与と不測死の関係性については、依然として不明である。成長ホルモン治療の開始に先立って行っておくことが推奨される評価については、「臨床的マネジメント」の項を参照されたい。
神経画像
脳画像でみられる異常としては、白質病変、脳室拡大、脳組織(頭頂葉・後頭葉)の体積減少、シルビウス裂多小脳回、島閉鎖不全、灰白質の体積の変化、下垂体高の減少などがある。これらのものと臨床症候との関連性は、今のところ不明である。
遺伝型-表現型相関
PWSに至る3つの主要な分子メカニズムのいずれについても、特定の表現型との関連は知られていない。ただ、いくつかの症候の出現頻度や重症度については、2大分子機構(15q欠失とUPD15)間で統計的な差がみられたとする報告がいくつかみられる。
- UPD15では、過期産[Butlerら2009]ならびに母親年齢の高まりがみられる[Millerら2011]。
- UPD15の罹患者については、特徴的顔貌、低色素沈着[Mahmoudら2021]、ジグソーパズルの能力[Dykens2022]といったものを有することが少ない。また、15q欠失例よりいくぶん言語性IQが高い[Rosenbergら2022]という違いもある。
- UPD15例のほうが、精神病[Yangら2013]、自閉症スペクトラム障害[Bennettら2015]を有する割合が高い。いくつかの研究によると、UPD15例では64%に非定型の精神病がみられたのに対し、15q欠失例では25%であったという[Yangら2013]。
浸透率
浸透率は100%である。
疾患名について
「HHHO」(性腺機能低下[hypogonadism],筋緊張低下[hypotonia],低知能[hypomentia],肥満[obesity])という名称は、現在はもはや用いられない。PWSは、時にWilli-Prader症候群、Prader-Labhart-Willi症候群とも呼ばれる。
発生頻度
多くの集団で、PWSの発生頻度は10,000人から30,000人に1人と推定されている。
16,579人の新生児に対してスクリーニングを行った最近のオーストラリアの研究では、8,290人に1人の発生頻度であった[Godlerら2022]。
遺伝学的に関連のある疾患
Angelman症候群は、PWS/ASクリティカル領域における母性発現の喪失に起因して生じる疾患である。これは、2歳以降、Prader-Willi症候群(PWS)とは臨床的に全く異なる経過をとる(「鑑別診断」の項を参照)。
Schaaf-Yang症候群は、15q11.2のPWSクリティカル領域にある父性継承のMAGEL2遺伝子に生じたトランケーション型病的バリアントにより引き起こされる疾患である。
母性継承のPWS/ASクリティカル領域の重複は、知的障害、てんかん発作、自閉症を引き起こす[Boyarら2001]。
「母性15q重複症候群」のGeneReviewを参照されたい。
鑑別診断
Prader-Willi症候群(PWS)と表現型の一部が類似する可能性のある疾患は多数に上る。
SNORD116欠失
PWSの表現型に関しては、SNORD116遺伝子クラスターの発現が失われることが1つの主要な役割を演じている(「分子レベルの病原」の項を参照)。SNORD116の欠失を有する稀な例が文献で報告されており、全部ではないものの、PWSの主要症候の多くがみられたとされている[Tanら2020]。SNORD116欠失例は、通常、PWSの顔面症候は有しないものの、その他の症候を軽度に有し、同時に、PWSでは通常みられない特徴(例えば、大頭症、成長ホルモン[GH]治療を行わない状態での高身長、多毛症、正常な思春期発達、正常知能)をいくつか併せもっていた。現在使用されているオリゴ-SNPコンビネーションアレイの大半で、SNORD116遺伝子クラスターの欠失が検出可能である点に注目されたい。
頭蓋咽頭腫
頭蓋咽頭腫、ならびにその治療後の状態は、PWSと大きく重なる部分がある。視床下部が損傷を受けると、PWSでみられる所見とほぼ同じ状況が現れるが、頭蓋咽頭腫が幼少期に生じたときは特にそれが言える。病歴を調べること、それでよくわからないときはDNAメチル化解析を行うことで、この疾患とPWSとの鑑別が可能である。
過食性低身長
過食性低身長は、社会心理的ストレスに起因して生じる後天性疾患で、成長ホルモン分泌不全、過食、軽度の学習障害などを伴う[Gilmourら2001]。病歴を調べること、それでよくわからないときはDNAメチル化解析を行うことで、この疾患とPWSとの鑑別が可能なはずである。
乳児期の筋緊張低下
乳児期の筋緊張低下は、新生児敗血症、中枢神経系の抑制、その他の遺伝性疾患(表4参照)でもみられる。これらの疾患については、PWSではほとんどみられない努力呼吸の減弱がみられることでPWSとの鑑別ができる場合がある。
表4:乳児期に筋緊張低下を呈しPrader-Willi症候群との鑑別を要する遺伝性疾患
| 疾患名 | 遺伝子 | 遺伝形式 | 臨床症候/コメント |
|---|---|---|---|
| Angelman症候群(AS) | UBE3A1 | メカニズムにより再発リスクが変わる2。 | 重度の発達遅滞あるいは知的障害,重度の言語障害,歩行失調ないし四肢の振戦,楽しげな態度・頻繁な笑い・微笑・易興奮性を伴う独特の行動。 小頭症とてんかん発作が多くみられる。 乳児期には筋緊張低下が唯一の症候である場合あり。 PWSでみられるような吸啜障害、性腺機能低下、顔貌の異常はみられない。 |
| 先天性筋無力症候群(CMS) | 以下を含む30超の遺伝子: CHAT CHRNE COL13A1 COLQ DOK7 RAPSN |
AR AD3 |
通常2歳未満で発症する眼・頭頸部・四肢の筋の易疲労性を伴う虚弱。 CMSは、古典的なものでは骨格筋のみに虚弱が現れる。 |
| 先天性筋強直性ジストロフィー | DMPK | AD | 出生時に筋緊張低下と重度の全身性筋虚弱がみられ、しばしば呼吸不全を伴って早期死亡に至る。 知的障害が多くみられる。 |
| 脆弱X症候群(「FMR1障害」のGeneReviewを参照) | FMR1 | XL | 罹患男性では中等度、罹患女性では軽度の知的障害がみられる。 男性には、特徴的外見、結合組織所見、巨大精巣(思春期後)がみられることあり。 行動異常が多くみられる。 乳児期には筋緊張低下が唯一の症候である場合あり。 PWSでみられるような吸啜障害、性腺機能低下、顔貌の異常はみられない。 |
| GARS1乳児発症型脊髄性筋萎縮症(「GARS1関連軸索型ニューロパチー」のGeneReviewを参照) | GARS1 | AD | 初期に現れる症候は、通常、呼吸窮迫、摂食不良、筋の虚弱(近位筋より遠位筋に顕著)である。 |
| 乳児発症型Pompe病 | GAA | AR | 筋緊張低下、全身性の筋虚弱、摂食障害、体重増加不良、呼吸窮迫、肥大型心筋症を伴う。 発症年齢の中央値は4ヵ月。 (注:PWSでは生後数ヵ月といった時期ではなく、出生段階で筋緊張低下が認められる。) |
| Schaaf-Yang症候群 | MAGEL2 | インプリンティングを伴うAD4 | 乳児期の筋緊張低下と摂食障害はPWSに似るが、PWSではみられないarthrogryposisなどの関節拘縮がみられる。 知的障害だけでなく自閉症スペクトラム障害もしばしばみられ、その程度は極度である場合あり。 小児期後期/思春期に肥満と過食を示し、年齢とともに顕著になる例あり。 |
| 脊髄性筋萎縮症 | SMN1 | AR | PWSではほとんどみられない努力呼吸の減弱がみられる場合あり。 |
| WAC関連知的障害 | WAC | AD | さまざまな程度の発達遅滞/知的障害あり。 年長の小児や成人の大半に行動異常がみられる。 大多数の乳児は、出生段階で、重度ながら非特異的な症候(例えば、新生児筋緊張低下,摂食障害)を示す。 |
| X連鎖性乳児型脊髄性筋萎縮症 | UBA1 | XL | 先天性の筋緊張低下、反射消失、ならびに脊髄・脳幹の前角細胞(すなわち、下位運動ニューロン)の変性や消失を示す症候が現れる。 しばしば先天性の拘縮や骨折がみられる。 |
| Zellwegerスペクトラム障害(ZSD) | ZSD-PEX遺伝子群 | AR5 | 罹患新生児は摂食不良を伴う筋緊張低下を示す。 特徴的顔貌、先天奇形、肝疾患(時に重篤な例あり)を有する。 (注:PWSでは脳MRI所見が比較的正常に近いことが多いのに対し、ZSDではきわめて顕著かつ特徴的な脳MRI所見がみられる。) |
AD=常染色体顕性;AR=常染色体潜性;XL=X連鎖性
- Angelman症候群(AS)は、母性継承のUBE3Aアレルの発現障害あるいは機能障害に起因して生じる疾患である。
- その発症メカニズムはいくつものものがある。
- Angelman症候群罹患者は、通常、孤発例(すなわち、家系内で1人だけの発生例)で、denovoの遺伝学的変化に起因して生じたものであることから、再発リスクはきわめて低い。ただ、比較的少数ながら、インプリンティングパターンに起因した遺伝学的変化の結果としてASが生じる場合もあり、その場合は常染色体顕性の遺伝形式をとり、再発リスクもさまざまな幅で現れることになる。
- 先天性筋無力症候群(CMS)は、通常、常染色体潜性の遺伝形式をとる。比較的少数ながら、常染色体顕性の遺伝形式をとるCMSもみられる。
- Schaaf-Yang症候群は、常染色体顕性遺伝のインプリンティング様式での継承を示す(父性由来のMAGEL2アレルに生じたヘテロ接合性病的バリアントにより、この疾患が生じる)。
- Zellwegerスペクトラム障害(ZSD)は、通常、常染色体潜性の遺伝形式をとる(PEX6の1バリアントであるp.Arg860Trpについては、ヘテロ接合状態でZSDが現れる)。
発達遅滞/知的障害と肥満(一部は性腺機能低下を伴う)
発達遅滞/知的障害と肥満を呈し、一部、性腺機能低下を伴う疾患群を表5にまとめて示した。表5:発達遅滞/知的障害と肥満(一部は性腺機能低下も)を呈し、Prader-Willi症候群との鑑別を要する遺伝性疾患
| 疾患名 | 遺伝子/遺伝学的メカニズム | 遺伝形式 | 臨床症候/コメント |
|---|---|---|---|
| Albright遺伝性骨ジストロフィー(「GNAS不活化障害」のGeneReviewを参照) | GNAS | インプリンティングを伴うAD1 | 低身長も特徴の1つであるが、筋緊張低下はみられず、PWSとは異なる特徴的顔貌(丸顔)を示す。 |
| Alstöm症候群 | ALMS1 | AR | 最初に現れる症候は、錐体-杆体ジストロフィーに起因する眼振、ないし、乳児発症型肥大型心筋症であることが多い。 その後に発症する所見としては、1歳未満で現れる肥満、進行性の感音性難聴、インスリン抵抗性/Ⅱ型糖尿病、思春期あるいは成人発症の拘束型心筋症、脂肪肝、進行性腎機能障害などがある。 |
| Angelman症候群(AS) | UBE3A2 | メカニズムにより再発リスクが変わる3。 | 15番染色体父性片親性ダイソミーにより生じたAS罹患者は、年齢の割に高いBMI値を示す。 大規模研究によると、70%超が過体重、40%超が肥満であった4。 |
| Bardet-Biedl症候群(BBS) | 以下をはじめとする26近い遺伝子: ARL6 BBS1 BBS2 BBS4 BBS10 BBS12 CEP290 MKKS5 |
AR | 錐体-杆体ジストロフィー,体幹型肥満,軸後性多指趾,高次脳機能障害,男性の低ゴナドトロピン性性腺機能低下,女性の複雑性器奇形,腎機能障害。 BBSとPWSでは、顔面の表現型が異なる。 |
| Borjeson-Forssman-Lehman症候群(BFLS)(OMIM301900) | PHF6 | XL | 男性では、重度の高次脳機能障害,てんかん,性腺機能低下,代謝低下,顕著な肥満,乳児期の筋緊張低下,発育不全。 PWSとは、知的障害の重症度、眼振の存在、ならびに、顕著な眼窩上隆起・眼瞼下垂・深い眼球といった顔貌の特徴で鑑別可能。 |
| Cohen症候群 | VPS13B | AR | 乳児期・小児期の体重増加不良,ティーンの年齢での体幹型肥満,早期発症型筋緊張低下と発達遅滞,1歳未満で始まる小頭症,中等度から極度の発達遅滞/知的障害,進行性網脈絡膜ジストロフィーと強度近視,多くの例で、反復性感染症と時にアフタ性潰瘍も伴った好中球減少症,明るい性格,関節過可動性。 PWSとは異なる特徴的顔貌がみられる。 |
| 脆弱X症候群(「FMR1障害」のGeneReviewを参照) | FMR1 | XL | この疾患の1つのサブセットにおいては、過食と肥満を含む「PWS様」表現型がみられる6。 |
| Prader-Willi症候群様症候群性肥満症7 | SIM1 | メカニズムにより再発リスクが変わる。 | 肥満と知的障害に加えて、筋緊張低下ないし四肢の短小化 |
| Temple症候群 (OMIM616222) |
母性片親性ダイソミー14,父性14番染色体欠失あるいは14q32の脱メチル化 | メカニズムにより再発リスクが変わる。 | 出生前の発育遅延、筋緊張低下、発達遅滞/知的障害を伴う乳児期の摂食障害、小児期の肥満、低身長は、Temple症候群とPWSに共通。 PWSとは、特徴的顔貌と思春期早発症で鑑別できる8。 |
AD=常染色体顕性;AR=常染色体潜性;XL=X連鎖性
- GNAS不活化障害は常染色体顕性遺伝を示し、その表現型は、問題のアレルがどちらの親起源かにより変わってくる。
- Angelman症候群(AS)は、母性継承のUBE3Aアレルの発現障害あるいは機能障害に起因して生じる疾患である。
- Angelman症候群罹患者は、通常、孤発例(すなわち、家系内で1人だけの発生例)で、denovoの遺伝学的変化に起因して生じたものであることから、再発リスクはきわめて低い。ただ、比較的少数ながら、インプリンティングパターンに起因した遺伝学的変化の結果としてASが生じる場合もあり、その場合は常染色体顕性の遺伝形式をとり、再発リスクもさまざまな幅で現れることになる。
- Lossieら[2001]
- 最も多くみられる遺伝子をここに挙げた。
- DeVriesら[1993]
- SIM1を含む6q16.2欠失の罹患者[Bonagliaら2008,ElKhattabiら2015]、ならびにSIM1の遺伝子内病的バリアントをもつ罹患者[Bonnefondら2013]で、「PWS様表現型」としての症候群性肥満が確認されている。
- Hosokiら[2009]
PWS類似の表現型を示す細胞遺伝学的異常
PWS類似の表現型を示す細胞遺伝学的異常としては、次のようなものがある。
- 6q16.3q23.3の重複(重複領域はSIM1を含まない)の1罹患者で、PWS様表現型が報告されている[Deschら2015]。
- 1p36の欠失によりPWS様表現型が現れるとの報告がいくつかみられる。
筋緊張低下、発達遅滞、肥満、過食、行動の問題がその所見である[Tsuyusakiら2010,Stagiら2014]。
- SHB2B1を含む16p11.2の欠失で、PWS様表現型が現れるとの報告が複数みられる。
SHB2B1は、レプチンシグナル伝達やインスリンシグナル伝達に関係する遺伝子である[Maillardら2015]。
- PWS様表現型が報告されているその他の細胞遺伝学的異常としては、dupXq27.2-ter、ならびにdel10q26がある[Lukusa&Fryns2000,Ben-Abdallah-Bouhjarら2012,Rocha&Paiva2014]。
臨床的マネジメント
Prader-Willi症候群(PWS)でみられる症候に対する管理は、年齢により変わってくる。
PWSの結果としてすでに表に現れたものだけでなく、今後現れることが予測される事態に関する情報提供までを含むものとする必要がある。チームアプローチが推奨され、すでに管理に関するアプローチがいくつか公表されている[McCandless2011,Cassidyら2012,Duisら2019]。これらに加え、国際Prader-Willi症候群機構(IPWSO)の臨床・学術諮問委員会のまとめた乳児期・幼児期・思春期・成人期それぞれにおける管理法の詳細が存在する。これについては、「IPWSOのドクター向けガイドライン」の中の「コンセンサス文書」で確認可能である。
最初の診断時に続いて行う評価
PWSと診断された罹患者については、疾患の範囲やニーズを把握するため、診断に至る過程ですでに実施済でなければ、表6にまとめた評価を行うことが推奨される。
表6:Prader-Willi症候群罹患者の最初の診断後に行うことが推奨される評価
| 系/懸念事項 | 評価 | コメント |
|---|---|---|
| 低下 |
|
|
| 発達遅滞 | 発達評価 |
|
| 性腺機能低下 |
|
|
| 内分泌 |
|
|
| 過食/肥満 |
|
|
| 行動 |
|
|
| 皮膚 | 皮膚むしり症に続発する開放創ないし感染に関する臨床的評価 | |
| 眼 | 眼科的評価 | 視力低下、異常眼球運動、斜視に関する評価を目的として行う。 |
| 睡眠 |
|
|
| 脊柱側彎 | 必要に応じ、臨床的評価とX線写真による評価 | 肥満度の強い例では脊柱側彎の臨床評価は難しく、診断の確定にはX線写真が必要。 |
| 股関節形成不全 |
|
|
| てんかん発作 | てんかん発作の懸念があるときは脳波を検討。 | |
| 歯 | 齲蝕リスクの上昇、歯の叢生、エナメル質形成不全に関する歯科的評価 | |
| 遺伝カウンセリング | 遺伝の専門医療職1の手で行う。 | 医学的、個人的な意思決定の用に資するべく、本人や家族に対し、PWSの本質、遺伝形式、そのもつ意味についての情報提供を行う。 |
| 家族への支援/情報資源 | 以下のニーズに関する評価
|
- 臨床遺伝医、認定遺伝カウンセラー、認定上級遺伝看護師をいう。
症候に対する治療
新生児科医に始まり、臨床遺伝医と遺伝カウンセラー、プライマリケア医、内分泌内科医、整形外科医、栄養士、心理士、精神科医、理学療法士、作業療法士、言語治療士、教育者へと続く多職種の専門家による管理が推奨される。
表7:Prader-Willi症候群罹患者の症候に対する治療
| 症候/懸念事項 | 治療 | 考慮事項/その他 |
|---|---|---|
| 乳児期の摂食障害 | 生後数週間から数ヵ月の間は、十分な栄養摂取と発育不全の回避を目的として、特殊乳首の使用、経鼻胃管での食物の強制摂取をはじめとする特殊な栄養補給が必要となる。 | PWSと診断された例は、経時的に摂食の問題が改善していくため、通常、胃瘻チューブが必要になることはない。 |
| 発達遅滞/知的障害 | 「発達遅滞/知的障害の管理に関する事項」の項を参照。 | |
| 性腺機能低下 |
|
|
| 思春期以降、内分泌内科医の手でホルモン補充療法を個別的に行う。 | 治療方針を検討する上では、骨粗鬆症や生殖能力/妊娠に関する可能性を頭に入れておくことが重要である。 | |
| 内分泌症候 |
|
|
| 内分泌内科医の指示に従い、思春期早発症、Ⅱ型糖尿病、甲状腺機能低下症、中枢性副腎不全の治療。 | ||
| 過食/肥満 | 食餌管理:
|
|
| 急性消化器症候 | 嘔吐ないし食欲不振は生命にかかわる疾病(胃炎あるいは胃壊死)の徴候である可能性があり、緊急入院ないし緊急手術が必要になることがある。 | 止瀉薬を使用することは、重篤な腸の膨満・壊死・破裂につながる可能性がある。 |
| 神経行動症候 |
|
|
| 皮膚症候 | 皮膚むしり症に対して、気持ちをそらせる、局所をカバーするといったことで対応できない場合、この問題の軽減あるいは除去を図る目的で、トピラマート(25-5mg/日)あるいはN-アセチルシステイン(450-1200mg/日)の使用を検討する4。 | 鼻出血や直腸出血がしばしば生じている例については、むしり症による皮膚損傷の可能性を迅速に評価する必要がある。 |
| 眼科的症候 | 眼科医による斜視ないし屈折異常の治療 | |
| 睡眠の問題 |
|
|
| 骨格の問題 | 一般集団と同様の形で行う脊柱側彎と股関節形成不全の治療 |
|
|
||
| てんかん発作 | 経験豊富な神経内科医の手で、抗痙攣薬を用いた標準治療を行う。 |
|
| 唾液分泌低下 | 特殊歯磨剤、歯磨きジェル、洗口剤、ガム等のドライマウスの治療用に開発された製品を用いた管理6。 | 生歯以降、3-4ヵ月に1度の歯科的評価と口腔衛生管理を検討する必要あり。 |
| 家族/地域社会 |
|
|
- Millerら[2006b]
- PWSの乳児・小児のカロリー必要量は、通常、1日推奨量の60%-80%である。PWS成人のエネルギー必要量が1,200-1,400kcal/日を超えることはほとんどない。
- Scheimannら[2012],Gantzら[2022]
- Bonnotら[2016]
- 両親/介護者に対して、多くみられるてんかん発作の現れ方に関する教育を行うことが望ましい。てんかんと診断された子どもに対する非医療的介入と対処の戦略に関する情報については、「EpilepsyfoundationToolbox」を参照されたい。
- Ritwik&Vu[2021]
発達遅滞/知的障害の管理に関する事項
以下に述べる内容は、アメリカにおける発達遅滞者、知的障害者の管理に関する一般的推奨事項を挙げたものである。ただ、そうした標準的推奨事項は、国ごとに異なったものになることもあろう。
0-3歳
作業療法、理学療法、言語治療、摂食治療、乳児のメンタルヘルスサービス、特別支援教育、感覚障害支援といったものが受けられるよう、早期介入プログラムへの紹介が推奨される。これは、アメリカでは連邦政府が費用を負担して、罹患者個人の治療上のニーズに対する在宅サービスが受けられる制度で、すべての州で利用可能である。
3-5歳
アメリカでは、地域の公立学区(訳注:ここで言う「学区」というのは、地理的な範囲を指す言葉ではなく、教育行政単位を指す言葉である)を通じて発達保育園に入ることが推奨される。入園前には、必要なサービスや治療の内容を決定するために必要な評価が行われ、その上で、運動、言語、社会性、認知等の機能の遅れをもとに認定された子どもに対し、個別教育計画(IEP)が策定される。通常は、早期介入プログラムがこうした移行を支援することになる。発達保育園は通園が基本であるが、医学的に不安定で通園ができない子どもに対しては、在宅サービスの提供が行われる。
全年齢
各地域、州、(アメリカの)教育関係部局が適切な形で関与できるよう、そして、良好な生活の質を最大限確保する支援を親に対してできるよう、発達小児科医とよく話をすることが推奨される。押さえておくべき事項がいくつかある。
- IEPのサービス
- IEPは、認定を受けた子どもに対して、特別に構成された指導と、それに関連するサービスを提供する。
- IEPのサービスは、必要な変更点を決定するため、毎年見直しが行われる。
- 特別支援教育に関する法律は、IEPの対象となっている子どもについて、いつ、いかなる場にあっても、学校という場で実現可能な範囲で最も制約の少ない環境に置かれるべきであり、適切と判断された範囲内で、可能な限り多くの一般教育が受けられる環境が与えられるべきであると規定している。
- 当該児のIEPチームには、子どもが教材に接するための支援を行う目的で、視覚に関する相談員が配置される必要がある。
- IEPにおいては、子どもが教材に接するために必要と思われる範囲内で、理学療法、作業療法、言語治療の提供が行われる。
それを超える部分については、罹患児のニーズに基づいて、自費での支援治療が検討されるようなこともある。
行う治療の種類については、発達小児科医が個別に推奨を行う。
- 子どもがティーン世代になった時点で、移行計画についての話し合いが行われ、IEPの中にそれが組み込まれる。
- IEPサービスを受ける人たちのため、公立学区はその人が21歳になるまでサービスを提供しなければならないことになっている。
- 504プラン(第504項:障害に基づく差別を禁じたアメリカの連邦法)に基づいて、教室内で前のほうへの着席、補助機器の使用、代書者の起用、業間の休み時間の延長、宿題の変更、大きな文字の教科書といった配慮や変更が必要な人に対しては、対応の検討が行われる。
- 発達障害者福祉局(DDA)への登録が推奨される。DDAは、アメリカの公的機関で、認定を受けた人に対してサービスや支援を提供している。認定基準は州によって異なるものの、ふつうは、診断名やそれに伴う認知/適応障害の度合に従って決定がなされる。
- 収入が少なく、情報資源も不足している家族については、障害児のための補足的所得補償制度(SSI)の認定を受ける道がある。
運動機能障害
粗大運動機能障害
- 可動性を最大限確保し、遅発性の整形外科的合併症(例えば、拘縮、脊柱側彎、股関節脱臼)のリスクを減少させることを目的として、理学療法が推奨される。
- 必要に応じて、耐久性医療機器や体位保持機器(例えば、車椅子、歩行器、バスチェア、補装具、障害者用ベビーカー)の使用を検討する。
- 筋緊張亢進やジストニアなど、筋緊張異常に対しては、バクロフェン、チザニジン、Botox®、抗パーキンソン病薬、整形外科的処置といったものによる対応を支援するため、当該分野を専門とする医療者との連携を検討する。
微細運動機能障害
摂食、身だしなみ、着替え、筆記などの適応機能に問題が生じる微細運動技能の障害に関しては、作業療法が推奨される。
口腔運動機能障害
口腔運動機能については、来院ごとに評価を行うようにする。そして、摂食時の窒息/嘔吐、体重増加不良、度重なる呼吸器疾患への罹患、特別な理由の見当たらない摂食拒否といった状況がみられる場合は、臨床的摂食評価やX線嚥下検査を行うようにする。罹患児が、口からの摂食を安全に行える状況にあるということが前提ではあるが、協調運動の改善、あるいは、感覚の関係した摂食の問題の改善を支援する手段として、摂食治療(ふつう、作業療法士あるいは言語治療士がこれを担当する)が推奨される。安全性を確保するため、食餌にとろみをつけたり、冷やしたりといったことが行われることがある。摂食機能障害が重度の場合は、経鼻胃管あるいは胃瘻チューブが必要になる場合がある。
コミュニケーションの問題
表出言語に障害をもつ罹患者に対しては、それに代わるコミュニケーション手段(例えば、拡大代替コミュニケーション[AAC])に向けての評価を検討する。AACに向けた評価は、その分野を専門とする言語治療士の手で行うことが可能である。この評価は、認知能力や感覚障害の状況を考慮に入れながら、最も適切なコミュニケーションの形を決めていこうというものである。AACの手段としては、絵カード交換式コミュニケーションシステムのようなローテクのものから、音声発生装置のようなハイテクのものまで、さまざまなものがある。一般に信じられていることとは反対に、AACはスピーチの発達を妨げるようなものではなく、むしろ理想的な言語発達に向けた支援を与えてくれるものである。
社会/行動上の懸念事項
小児に対しては、応用行動分析(ABA)をはじめとする自閉症スペクトラム障害の治療で用いられる治療的介入の導入に向けた評価を行うとともに、実際にそれを施行することがある。ABA療法は、個々の子どもの行動上の強みと弱み、社会性に関する強みと弱み、適応性に関する強みと弱みに焦点を当てたもので、ふつう、行動分析に関する学会認定士との1対1の場で行われる。
発達小児科医を受診することで、両親に対し、適切な行動管理の指針を指導したり、必要に応じ、強迫性の症候や精神病の治療薬を処方したりといったことも可能になる。
深刻な攻撃的、破壊的行動に関して懸念があるときは、小児精神科医への相談という形での対応が考えられる。
定期的追跡評価
アメリカ小児科学会(AAP)から、健康管理のガイドラインが公表されている[McCandless2011](全文はこちら)。最近、PWSに係わる専門家グループの調査と文献レビューに基づいて、ケアに向けた集学的アプローチが公表されている[Duisら2019]。
表8:Prader-Willi症候群罹患者で推奨される定期的追跡評価
| 系/懸念事項 | 評価 | 実施頻度 |
|---|---|---|
| 発達遅滞/知的障害 | 発達の進行状況と教育上のニーズに関するモニタリング。 | 来院ごと。 |
| 停留精巣 | 精巣固定術の後、停留精巣が再発することがあるため、精巣の位置のモニタリング。 | 年に1度。 |
| 内分泌 | 糖尿病に関する評価を目的に、糖化ヘモグロビン濃度の評価とブドウ糖負荷試験。 | 肥満の場合は年に1度、もしくは思春期から始め年に1度。 あるいは急激な体重増加その他の症候がみられた場合。 |
| 甲状腺機能低下の評価を目的に、遊離T4、甲状腺刺激ホルモン値の評価。 | 乳児期から始め6-12ヵ月ごと。 | |
| 中枢性副腎不全に関する評価。 | 症候に基づいた必要性に応じて、ならびに病気や手術に際して。 | |
| 成長/食欲/肥満 | 身長、体重、BMI1のモニタリング。 |
|
| 行動/精神 | 家族・介護者とともに、行動の所見や強迫性の症候の有無を評価。 | 2歳以降、年に1度。 |
|
思春期と成人期に年に1度。 | |
| 皮膚 | 傷や、皮膚の感染の徴候/症候の有無に関する診査。 | 来院ごと。 |
| 視覚 | 斜視や屈折異常の評価を目的とした眼科的検査。 | 年に1度。 |
| 睡眠 | 家族・介護者とともに、いびき、夜間の頻回の覚醒、新たな行動の問題に関する評価。 | 年に1度。 |
| 睡眠検査による睡眠中の呼吸の状態の評価。 | 成長ホルモン治療開始前と開始後4-8週、ならびにその後、臨床症候(いびき、夜間の頻回の覚醒、行動の問題)が現れたとき。 | |
| 終夜睡眠検査に続いて、複数の睡眠潜時検査とビデオ脳波。 | 日中の過度の眠気/ナルコレプシー/カタプレキシーを有する例について行う。 | |
| 脊柱側彎 | 脊柱側彎に関する臨床的診査。 | ひとり座りができるようになって以降、来院ごと。 |
以下を有する例については、脊柱側彎を調べるための脊椎のX線写真。
|
年に1度。 | |
| 骨粗鬆症 | DXA法による骨密度測定。 | 思春期から始め、2年に1度。 |
| てんかん発作 | 発作を有する例について、新たな発作に関する評価と、臨床的必要性に応じたモニタリング。 | 来院ごと。 |
| 歯 | 歯科的評価。 | 生歯以降少なくとも6ヵ月ごと。 歯科的問題を有する例については3-4ヵ月ごと。 |
| 家族/地域社会 | 家族に対するソーシャルワーカーの支援(例えば、緩和/息抜きケア,在宅介護,その他の地域の情報資源)の必要性、ケアコーディネーションの必要性、ならびに新たな質問(例えば、家族計画)が生じた場合のフォローアップの遺伝カウンセリングの必要性に関する評価。 | 来院ごと。 |
1.体重は㎏で、身長はmの2乗で計算。
避けるべき薬剤/環境
自由に食物をとれる環境
過食は、早食いとそれに伴う致命的な窒息、不顕性誤嚥、腐敗した食物の摂取につながるおそれがあるため、監視の目なしで食物に近づける状況は避ける必要がある。
催吐薬
未調理あるいは腐敗した食物を摂ってしまった後、それを吐き出させる上では、催吐薬は通常無効で、反復して服用することによる毒性の可能性がある。それに代わり、胃の減圧を目的として経鼻胃管を使用するようにすべきである。
止瀉薬
PWS罹患者については、止瀉薬が腸の重度の膨満、壊死、破裂につながるおそれがあることから、これを避ける必要がある。腹部の膨満、鼓脹、疼痛、不活動、食欲喪失、嘔吐といったものは、生命を脅かすような胃の炎症あるいは壊死の徴候である場合がある。こうした症候を有する例については、医療機関での評価を迅速に行い、場合によっては入院させる必要がある。放射線による画像診断とともに、緊急手術を要する可能性も考えられる。
鎮静剤その他の薬剤
PWS罹患者は、標準的用量の投薬に対して異常反応を示す可能性がある。薬剤、特に鎮静効果をもつ薬剤については、異常反応が長く続いたとする報告があることから、投与は慎重の上にも慎重に行うべきである。肥満についても、適切な投与量に影響してくる可能性が考えられる。
リスクを有する血縁者の評価
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
妊娠に関する管理
PWS女性の妊娠は稀である。PWS罹患者、中でも肥満を抱えるPWS罹患者は疼痛閾値が高いため、妊娠に際しては、より頻回のモニタリングが必要である。
研究段階の治療
現段階で、過食に対する治療法として最も有望なデータがあるのは、diazoxidecholinecontrolledrelease(DCCR)の試験で報告されているものである。DCCRは、ATP感受性K(K-ATP)チャンネル作動薬で、膵β細胞からのインスリン分泌に対する下向き調節、神経ペプチドY分泌の下向き調節、脂肪組織中のK-ATPチャンネル活性化などを通じて、PWS罹患者に有益な効果を発揮する可能性を有している。DCCRでは、1年間の治療により、過食質問票スコア上で9ポイントを上回る改善を示している。現在進行中の試験のデータでは、体組成の改善(DXAスキャンでみたときの除脂肪体重値ならびに除脂肪筋肉量対脂肪比)に加え、PWSでみられる特徴的な行動の問題の多く(例えば、質問の繰り返し、不安、強迫行動、皮膚むしり症)で改善がみられている[Millerら2023]。この治療法は、現在、FDAによる審査を受けている段階である。
過食の抑制については、カルベトシン(合成オキシトシンアナログ)も一定の効果を示すことがわかっている。カルベトシンは、アルギニンバゾプレッシンの活性化抑制を狙って設計されたオキシトシン受容体特異的な化合物である。この薬剤の第Ⅱ相臨床試験では、2週間の投与で、過食質問票スコアの低下という有望な結果が得られている[Dykensら2018]。
最近終了した低用量カルベトシン(3.2mg)を用いた第Ⅲ相臨床試験では、過食質問票スコアの3.1ポイントの改善とともに、強迫性行動や不安に関する改善もみられている[Roofら2023]。この新薬の申請は、2021年11月に却下され、臨床試験の追加が必要との指摘を受けた経緯がある。
PWS罹患者の過食に対する治療薬としては、その他にも、オキシトシン、カンナビジバリン、メラニン凝集ホルモン受容体1作動薬、腸の苦み受容体活性化化合物、カンナビジオールなど、いくつかの第Ⅱ相臨床試験が進行中である。
日中の過度の眠気やナルコレプシー/カタプレキシーに対する治療薬としては、ピトリサントの第Ⅱ相臨床試験が進行中である。
経皮的迷走神経刺激では、PWSの成人を対象とした小規模試験で感情爆発の頻度と強度を減少させる有意の効果がみられている。効果の再現性と持続時間を明らかにすることを目的として、現在、より大規模なプラセボ対照試験が計画されているところである[Manningら2019]。
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「ClinicalTrials.gov」、ならびにヨーロッパの「EUClinicalTrialsRegister」を参照されたい。PWS関連の臨床試験については、これらに加え、Mahmoudら[2023]による詳細なレビューも参照されたい。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
Prader-Willi症候群(PWS)罹患者は、通常、孤発例(すなわち、家系内で1人だけの発生例)で、denovoの遺伝学的変化に起因して本疾患に至った例である。大多数の家系について言うと、再発リスクは1%未満であるが、病因によっては、再発リスクが50%になることもあり、また、ふつうはありえないこととはいえ、シナリオ(すなわち、母親が15/15のロバートソン転座)によっては再発リスクがほぼ100%になるようなことも理論上はありうる。そのため、PWSの再発リスク評価を信頼性のあるものとするためには、発端者でPWSが生じた遺伝学的メカニズム(すなわち、15q欠失,15番染色体の片親性ダイソミー[UPD15],インプリンティング障害)とその背景にある遺伝学的病因を明らかにするとともに、両親に対して遺伝学的変化が生じる素因(すなわち、片親の有する染色体再構成、あるいは父親がインプリンティングセンターの欠失をヘテロで有していること)の有無を調べる検査を行うことが必要となる。
家族構成員のリスク
発端者の両親
発端者の両親は、ともに非罹患者である。
発端者の同胞
- PWS発端者の同胞の有するリスクは、発端者のPWS発症の背景にある遺伝的メカニズムと遺伝学的病因、ならびに両親の遺伝学的状態によって変わってくる(表9ならびに表10を参照)。
- 15q11.2-q13領域のDNAメチル化異常の同定、ならびにオリゴ-SNPコンビネーションアレイ(OSA)による背景としての遺伝学的メカニズム(15qの欠失,母性UPD15,あるいはインプリンティング障害)の同定がなされ、PWSの診断が確定したら、再発リスク評価のための遺伝学的病因の確定を進める必要がある。発端者における遺伝学的病因、ならびに両親の遺伝学的状態を確認するために推奨される検査を、表9と表10にまとめて示した(表1も参照のこと)。
表9:遺伝学的メカニズム別にみたPrader-Willi症候群発端者の同胞の有するリスク
| 遺伝学的メカニズム | 考えられる遺伝学的病因 | PWSに占める割合 | 遺伝的病因の判別ならびに素因としての親の遺伝学的変化の有無確認のために推奨される検査 | 同胞の有するリスク |
|---|---|---|---|---|
| 15q欠失 | denovoの欠失 | 60%-70% |
|
1%未満2 |
| 不均衡型染色体再構成に起因する欠失 | 1%未満 | 最大25% | ||
| UPD15 | denovoのUPD15 | 30%-40%近く | 発端者:核型解析
|
1%未満 |
| 素因となる染色体異常(例えば、父性の15/15ロバートソン転座,マーカー染色体)を伴うUPD15 | 1%未満 | 1%未満から100%まで4 | ||
| インプリンティングセンター(IC)の欠失 | denovo | 0.5%未満 |
|
1%未満2 |
| 父由来のIC欠失 | 0.5%未満5 | 50% | ||
| エピ変異によるインプリンティング障害 | denovo | 2%-4% | 追加検査なし | 1%未満 |
MS-MLPA=メチル化特異的MLPA法;OSA=オリゴ-少塩基多型(SNP)コンビネーションアレイ;UPD=片親性ダイソミー
- オリゴ-SNPコンビネーションアレイ(OSA)では、15q近位に生じた転座や逆位は検出されない。
- 父親の生殖細胞系列モザイクは稀であるが、15q11.2欠失[Kokkonen&Leisti2000,Fernández-Novoaら2001]ならびにIC欠失[Buitingら2003,Weyら2005,DDriscollの個人的観察]について実例が存在する。
- 父親が15/15ロバートソン転座を有している場合は、減数第一分裂における異常分離により零染色体精子が生じることになる。これとモノソミーレスキューが組み合わさることでダイソミーが生じ、胚は15番染色体母性片親性アイソダイソミーをもつに至る。
- きわめて高い再発リスクも理論上はありうることながら、経験的に言うと、ほとんどの場合、再発リスクは1%未満であるように思われる。
- インプリンティングセンター(IC)の欠失の半数は父親からの継承であり、残る半数はdenovoである。
表10:遺伝学的メカニズム別、遺伝学的病因別にみたPrader-Willi症候群発端者の同胞の有するリスク
| 遺伝学的メカニズム | 遺伝的病因の判別ならびに素因としての親の遺伝学的変化の有無確認のために推奨される検査 | 遺伝学的病因 | PWSに占める割合 | 同胞の有するリスク |
|---|---|---|---|---|
| 15q欠失 |
|
denovoの欠失 | 60%-70% | 1%未満2 |
| 不均衡型染色体再構成に起因する欠失 | 1%未満 | 最大25% | ||
| UPD15 | 発端者:核型解析
|
denovoのUPD15 | 30%-40%近く | 1%未満 |
| 素因となる染色体異常(例えば、父性の15/15ロバートソン転座,マーカー染色体)を伴うUPD15 | 1%未満 | 1%未満から100%まで4 | ||
| インプリンティングセンター(IC)の欠失 |
|
denovo | 0.5%未満 | 1%未満2 |
| 父由来のIC欠失 | 0.5%未満5 | 50% | ||
| エピ変異によるインプリンティング障害 | 追加検査なし | denovo | 2%-4%近く | 1%未満 |
MS-MLPA=メチル化特異的MLPA法;OSA=オリゴ-少塩基多型(SNP)コンビネーションアレイ;UPD=片親性ダイソミー
- オリゴ-SNPコンビネーションアレイ(OSA)では、15q近位に生じた転座や逆位は検出されない。
- 父親の生殖細胞系列モザイクは稀であるが、15q11.2欠失[Kokkonen&Leisti2000,Fernández-Novoaら2001]ならびにインプリンティングセンター(IC)欠失[Buitingら2003,Weyら2005,DDriscollの個人的観察]について実例が存在する。
- 父親が15/15ロバートソン転座を有している場合は、減数第一分裂における異常分離により零染色体精子が生じることになる。これとモノソミーレスキューが組み合わさることでダイソミーが生じ、胚は15番染色体母性片親性アイソダイソミーをもつに至る。
- きわめて高い再発リスクも理論上はありうることながら、経験的に言うと、ほとんどの場合、再発リスクは1%未満であるように思われる。
- IC欠失の半数は父親からの継承であり、残る半数はdenovoである。
発端者の子
- 女性で例外が稀にみられるものの、PWS罹患者が生殖に至ることはない。遺伝学的にPWSの確定がなされた男性については、生殖に至ったとする報告はこれまでみられない。
- 子の有するリスクは、正式な遺伝カウンセリングの中で判断する必要がある。
他の家族構成員
発端者とその片親に染色体再構成が同定された場合には、保因者である片親の同胞に対し、遺伝カウンセリングを行うとともに、遺伝学的検査の選択肢を提供する必要がある。
- 発端者の父親がインプリンティングセンターの欠失をヘテロで有していた場合には、父親の同胞についても、そのインプリンティングセンターの欠失を有するリスクがあることになる。
関連する遺伝カウンセリング上の諸事項
家族計画
- 遺伝学的リスクの確定、出生前/着床前遺伝学的検査を受けるかどうかの話し合いといったことに最も適しているのは、妊娠前の時期である。
- 罹患者の両親に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
PWSの子をもつ家族
発端者において原因となった遺伝学的メカニズムが特定された場合には、PWSに関する出生前検査を行うことが可能となる[Butler2017]。出生前検査に向けてのアプローチは、発端者で同定された分子メカニズムの内容によって変わってくるが、PWSでみられる3種の分子的原因のいずれについても、出生前検査が可能である。出生前の組織についてインプリンティング障害を同定する上で、5’SNRPNの座位のDNAメチル化解析は最も信頼に足る手法であることに注目されたい[Glennら2000,Beygoら2019]。
PWSの家族歴を有しない妊娠
以下の状況にある場合は、PWSが生じる可能性がある。
- 絨毛膜絨毛サンプリング(CVS)もしくは羊水穿刺で得られた細胞を用いた細胞遺伝学的検査で15q11.2の欠失が疑われる場合には、OSAが行われることになる。その場合、欠失の確認を行った後、その欠失が母親由来(胎児はAngelman症候群)か父親由来(胎児はPrader-Willi症候群)かを確かめるため、起源となった親を調べる検査(parent-of-originstudy)を行う必要がある。
- CVSで得られた細胞の検査で、15トリソミーもしくは15トリソミーモザイクが検出され、かつ、羊水穿刺で得られた細胞のその後の検査で染色体数が46本であることがわかった場合には、トリソミーレスキューにより母性15番染色体が失われてAngelman症候群(父性UPD)に至った可能性、あるいは、父性15番染色体が失われてPrader-Willi症候群(母性UPD)に至った可能性を検討する必要がある。
この場合は、羊膜細胞を用いたparent-of-origin(UPD)検査もしくはDNAメチル化解析を検討すべきである。
- 継承あるいはdenovoの形で15番染色体が関与する転座がみられる場合、あるいは、15番染色体由来の過剰染色体がみられる場合は、OSA(欠失の可能性排除が目的)に加えて、parent-of-origin検査あるいはDNAメチル化解析(UPDの可能性排除が目的)を行うことになる。
非侵襲的出生前検査(NIPT)
胎児セルフリーDNAを用いた非侵襲的出生前検査で、15q11.2の欠失を調べることも可能であるが、これは偽陽性率が高い(主要な13・18・21トリソミーの検査は高い感度・特異度を示すが、これとは偽陽性率が異なる)。また、NIPTではASの欠失とPWSの欠失を判別することはできず、UPDやインプリンティング異常の検出もできない。
着床前遺伝学的検査(PGT)
インプリンティングセンターの欠失が判明した一部の家系については、着床前遺伝学的検査が1つの選択肢となりうる。PGTはまた、家族性の転座を有する例に対して、UPDの可能性を排除する目的で使用することも可能である。
出生前検査の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。
現在、多くの医療機関では、出生前検査を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- InternationalPrader-WilliSyndromeOrganisation(IPWSO)
- InternationalPrader-WilliSyndromeOrganisation:MembersAroundtheWorld
- MedicalHomePortal
- Prader-WilliSyndromeAssociationUSA
- FoundationforPrader-WilliResearch
- Prader-WilliSyndromeAssociationUK
SalisburyHouse
StationRoad
UnitedKingdom
Email:info@ipwso.org
www.ipwso.org
OurmembersareconstitutedPWSAssociationsrepresentingfamiliesaroundtheworld.
PWS Associations
Phone:941-312-0400
www.pwsausa.org
Phone:888-322-5487
Email:info@fpwr.org
www.fpwr.org
UnitedKingdom
Phone:+44(0)1332365676
Email:admin@pwsa.co.uk
pwsa.co.uk
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:Prader-Willi症候群:遺伝子とデータベース
| クリティカル領域 | 遺伝子 | 染色体上の座位 | タンパク質 | ClinVar |
|---|---|---|---|---|
| PWCR | 不明 | 15q11.2 | 不明 |
データは、以下の標準資料から作成したものである。遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:Prader-Willi症候群関連のOMIMエントリー(内容の閲覧はOMIMへ)
| 137142 | GAMMA-AMINOBUTYRIC ACID RECEPTOR, ALPHA-5; GABRA5 |
| 137192 | GAMMA-AMINOBUTYRIC ACID RECEPTOR, BETA-3; GABRB3 |
| 176270 | PRADER-WILLI SYNDROME; PWS |
| 182279 | SMALL NUCLEAR RIBONUCLEOPROTEIN POLYPEPTIDE N; SNRPN |
| 600161 | PRADER-WILLI/ANGELMAN REGION RNA 1; PWAR1 |
| 600233 | GAMMA-AMINOBUTYRIC ACID RECEPTOR, GAMMA-3; GABRG3 |
| 601491 | IMPRINTED IN PRADER-WILLI SYNDROME; IPW |
| 601623 | UBIQUITIN-PROTEIN LIGASE E3A; UBE3A |
| 602117 | NECDIN; NDN |
| 603856 | MAKORIN 3; MKRN3 |
| 603857 | MKRN3 ANTISENSE RNA; MKRN3AS |
| 605283 | MAGE-LIKE 2; MAGEL2 |
| 605436 | SMALL NUCLEOLAR RNA, C/D BOX, 116-1; SNORD116-1 |
| 605837 | HECT DOMAIN AND RCC1-LIKE DOMAIN 2; HERC2 |
| 605855 | ATPase, CLASS V, TYPE 10A; ATP10A |
| 609837 | SMALL NUCLEOLAR RNA, C/D BOX, 115-1; SNORD115-1 |
| 610922 | NUCLEAR PORE ASSOCIATED PROTEIN 1; NPAP1 |
| 611215 | PRADER-WILLI REGION NONCODING RNA 1; PWRN1 |
| 611409 | OCA2 GENE |
分子レベルの病原
Prader-Willi症候群(PWS)クリティカル領域(PWCR)は、15番染色体長腕近位の5-6Mbのゲノム領域に局在する(図2参照)。その領域内にあるこれより小さな2.5Mbのインプリンティング可変領域がそれである。PWSは一種の隣接遺伝子疾患で、症候がすべて揃った完全な表現型は、数個の遺伝子の発現がなくなることで生じることが、これまでの研究で明らかになっている[Cassidyら2012]。ただ、表現型形成の上では、SNORD116遺伝子クラスターの発現停止が主要な役割を演じていることがわかってきている[Tanら2020]。
15q11.2-q13領域にある関連遺伝子群の発現は、どちらの親由来かによって決まっていることから、PWSはまた、インプリンティング疾患でもある[Cassidyら2012]。
PWSを引き起こすゲノムやエピゲノムの変化は、すべて、15q11.2-q13にある本来父性発現の遺伝子の発現を失わせるものである。こうした遺伝子の父親由来のコピーが失われたり、発現できなくなったりすることは、取りも直さず、罹患者ではこれらの遺伝子の発現が完全に失われることを意味する。なぜなら、母親由来の遺伝子については、エピジェネティックな要因によってサイレンシングのプログラミングがなされているからである[Cassidy&Driscoll2009,Cassidyら2012]。
欠失のメカニズム
15q11.2-q13中間部の父由来アレルの微小欠失は、PWS罹患者の60%-70%を占める。
欠失の大多数は、近位の頻出切断点BP1、BP2のいずれか1つと、遠位の頻出切断点BP3との間で生じ、5Mbから6Mbの欠失に至るものである(図2参照)。頻出切断点(BP1,BP2,BP3)の近傍には、複数のタンデムリピートが存在する。減数分裂に際して、これら250-400kbの低コピー反復配列の部分で非相同性の対合が生じ、15番染色体に欠失(どちらの親由来かによって、PWS、Angelman症候群[AS]のどちらかが生じる)、重複(母性・父性の両方がある)、三重化(triplication)、逆位重複といったものが生じる結果となる。PWS罹患者の約8%は、頻出切断点以外の部位に生じた非定型サイズの欠失を示す。これは、不均衡型転座をはじめとするさまざまな要因で生じる[Kimら2012,Butlerら2019a]。非定型の欠失を有する例は、定型のPWS罹患者よりも臨床症候が軽度である場合もあれば重度である場合もある。
インプリンティングセンター欠失
母親特異的メチル化パターンを示すものの、15q11.2-q13の欠失も母性片親性ダイソミー(UPD)も有しない例の中に、プロモーター領域とSNRPNの近位上流領域(インプリンティング調節エレメントと目される部分がここに含まれている)の小欠失が確認されている例が複数報告されている。
また、二親性の継承を有しつつも、この領域が母性メチル化パターンしか示さず、インプリンティングセンターにも検出しうる異常がみられない例が複数みられる。こうした例は、エピ変異に起因するインプリンティング障害と考えられる。
この領域内にある主な遺伝子
PWS/ASクリティカル領域には、次のような遺伝子がマッピングされている(図2参照)。
- ATP10A
ATP10Aは、もともと母性優先発現遺伝子であると考えられてきたが、その後のマウス[DuBoseら2010]ならびにヒト[Hogartら2008]の研究で、これがインプリンティングを受けた遺伝子であることが疑問視されるようになった。そうではなく、ランダムなモノアレル性発現であるとする見方がある[Hogartら2008]。
- GABRB3,GABRA5,GABRG3
これらは、脳内の神経伝達に関与するGABA受容体サブユニット関連の遺伝子である。
- HERC2
HERC2は核-細胞質間を行き来して、標的タンパク質のユビキチン化と分解に必要なE3ユビキチンリガーゼの1つとして、他のE3ユビキチンリガーゼの活性化因子として、そしてまた、DNA損傷応答タンパク質会合のためのアダプターとして機能している。OCA2からHERC2にかけての座位は、ヒトにおいて眼の色を決める最大の要素となっている[Suarezら2021]。この領域を跨ぐ欠失を有するPWS罹患者が家族内の他の人に比べ概して低色素であることの背景には、こうしたことが関係しているものと思われる。
- IPW
IPWはSNORD116-1とSNORD115-1の間に位置する長いノンコーディングRNAである。これは、これまでに、14番染色体にあるDLK1-DIO3インプリント領域の調節因子であることがわかっており、15番染色体以外の調節に関与している模様である[Stelzerら2014]。PWSの中で果たしている役割は不明であるが、SNORD116の欠失を有する7例がすべてこの座位を含む欠失であり、しかもこの7例がPWSの主要症候の多くを有しているという事実は興味深いところである[Tanら2020]。
- MAGEL2
MAGEL2の転写は、父性アレルのみから行われ、主として脳内で発現する。
MAGEL2の父性アレルに機能喪失型バリアントを有する例は、Schaaf-Yang症候群となって現れる。これは、PWSと複数の症候が重なる疾患である。
- MKRN3
MKRN3は、父性の染色体からのみ発現するジンクフィンガータンパク質の1つをコードしている。ヒトにおいては、MKRN3の父性継承の機能喪失型バリアントにより、家族性中枢性思春期早発症が生じる[Abreuら2013,Macedoら2014]。
- NDN
NDNは父性発現遺伝子で、神経系の発生初期において抗アポトーシス因子(生存因子)として機能するDNA結合タンパク質の1つをコードしている[Andrieuら2006]。そしてこれは、ゴナドトロピン放出ホルモンニューロン内で一定の役割を果たしている可能性がある[Chungら2020]。
- NPAP1
NPAP1はイントロンをもたない遺伝子で、成人の精巣内では両アレル性の発現を示すが、胎児の脳内では片アレル性の発現である。
- OCA2
OCA2はメラノソームタンパク質の1つをコードし、この遺伝子の病的バリアントは眼皮膚白皮症2型を引き起こす。また、PWS罹患者において多くみられる低色素沈着は、この遺伝子の欠失と関連がある。
- PWRN1
PWRN1は精巣で発現し、これより低レベルながら、前立腺、心臓、腎臓、肝臓、肺、骨格筋、気管、脊髄、胎児の脳といったものにおいても発現していることが確認されている。なお、胎児の脳内での発現は片アレル性であることが明らかになっている。
- SNORD115(snoRNAHBII-52)
SNORD115は、boxC/D型核内低分子RNA(snoRNA)のクラスターで、おおむね47コピーで構成される。これは専ら神経細胞内でのみ発現し、また父性のみの発現を示す。SNORD115は、セロトニン2c受容体の生成を促進していることが示唆されている。ただ、稀ではあるが、この座位を含まない欠失を有するPWS罹患者も存在する。そうしたことから、これはPWSの表現型を形成する主要要素ではないものの、この部分の欠失がPWSの行動面での表現型のいくつかに関与している可能性はありうるように思われる[Chungら2020]。
- SNORD116(snoRNAHBII-85)
おおむね24コピーから成るsnoRNASNORD116クラスターがPWSの主要症候の中の多くのものの原因になっていることを、データから読み取ることができる。SNORD116の欠失という稀な例が数症例報告されており、これらはPWSの主要症候の多くを具備していたという[Tanら2020]。snoRNAの遺伝子はおそらく、選択的スプライシングを介したmRNAの修飾に関与しているものと思われ、1つ1つのsnoRNAがそれぞれ複数の標的を有している可能性がある。ただ、SNORD116に関しては、今のところ確定的な標的の同定はなされていない。
- SNURF-SNRPN
SNURF-SNPRN(ふつうは単にSNPRNと表記される)は、2つの別々のタンパク質をコードする複雑なバイシストロン性遺伝子の1つである。エクソン4-10が最初に解明されたが、これは、mRNAのスプライシングに関与するスプライセオソームのタンパク質の1つであるSmNをコードしている。SNURFは、エクソン1-3によってコードされ、まだ機能がよくわかっていないポリペプチドを産生する。SNURFは同時に、テロメア側に位置する7つのsnoRNA遺伝子の宿主遺伝子としての働きもしており、SNURF-SNPRNの発現によってこれらの調節が行われる仕組みになっている[Chungら2020]。
- UBE3A
UBE3Aは、脳内でのタンパク質の分解に関与するE3ユビキチンリガーゼで、ASの発生に関係している。
この領域内にあるインプリンティング遺伝子
Prader-Williクリティカル領域(PWCR)内にある数多くの遺伝子(MKRN3,MAGEL2,NDN,NPAP1,PWRN1,SNORD116,IPW,SNORD115,SNURF-SNRPN)がゲノムインプリンティングの対象となっており、このことは、父親由来のPWCRが失われたときのみにPWSの表現型が現れるということをよく表している。ゲノムインプリンティングのプロセスにはDNAのメチル化が係わっているが、これまでに、PWCR内にある遺伝子のいくつかにこのメチル化が生じていることが明らかになっている[Cassidyら2012,Beygoら2019,Chungら2020]。PWSの中のほんの数例ではあるが、母親特異的DNAメチル化パターンを示すものの、多くみられる父親由来のPWS/ASクリティカル領域の大欠失も母性片親性ダイソミーもみられず、ただSNRPNの上流にあるインプリンティング調節エレメントと目される領域の微小欠失が認めるのみという例が存在する[Cassidyら2012,Beygoら2019]。それ以外に、エピ変異による散発性のインプリンティング異常が明らかになった例も存在する[Beygoら2019]。
更新履歴:
- Gene Review著者: Suzanne B Cassidy,MD,FACMG,FAAP, Stuart Schwartz,PhD,FACMG.
日本語訳者: 窪田美穂(ボランティア翻訳者),鳴海洋子(信州大学医学部附属病院遺伝子診療部)
Gene Review 最終更新日: : 2008.3.24. 日本語訳最終更新日:2009.5.31. - Gene Review著者: Daniel J Driscoll, MD, PhD, FACMG, FAAP, Jennifer L Miller, MD, MS, FAAP, Stuart Schwartz, PhD, FACMG, and Suzanne B Cassidy, MD, FACMG, FAAP.
日本語訳者: 江田 肖(瀬戸病院 遺伝診療科) ,櫻井晃洋(札幌医科大学遺伝医学)
Gene Review 最終更新日: 2014.1.23 日本語訳最終更新日: 20140.10.28 -
Gene Reviews著者: Daniel J Driscoll, MD, PhD, FFACMGG, FAAP, Jennifer L Miller, MD, MS, FAAP, and Suzanne B Cassidy, MD, FFACMGG.
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2023.3.9. 日本語訳最終更新日: 2023.7.30.[in present]
![]()