Angelman症候群(アンジェルマン症候群)
(Angelman Syndrome)
Gene Reviews著者: Aditi I Dagli, MD,Jennifer Mathews, MS, CGC,and Charles A Williams, MD.
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2021.4.22. 日本語訳最終更新日: 2023.7.30.
要約
疾患の特徴
Angelman症候群(AS)は、重度の発達遅滞や知的障害、重度の言語障害、失調性歩行ないし四肢の振戦、頻繁な笑い・ほほ笑み・興奮等の見るからに愉快そうな振る舞いを伴う独特の行動を特徴とする疾患である。小頭症とてんかん発作も多くみられる。発達遅滞が初めて確認されるのは生後6ヵ月前後であるが、ASの独特の臨床症候が顕在化するのは1歳を過ぎてからである。
診断・検査
発端者におけるASの診断は、コンセンサスを得た臨床診断基準を満たすこと、ないしは、分子遺伝学的検査にてUBE3Aの母親由来アレルの発現低下や機能低下を示唆する所見が得られることをもって確定する。15q11.2-q13領域の片親特異的DNAメチル化インプリンティングの解析により、AS罹患者の約80%で、欠失、片親性ダイソミー、インプリンティング異常などが検出される。細胞遺伝学的に視認可能な染色体再構成(例えば、転座や逆位)を有する例が1%未満存在する。さらに、UBE3Aの配列解析で、約11%の罹患者に病的バリアントが検出される。すなわち、分子遺伝学的検査(メチル化解析とUBE3Aの配列解析)で罹患者の約90%に何らかの変化が確認されるということになる。ASの古典的表現型を有する残り10%の罹患者は、今もって特定に至っていない何らかの遺伝学的メカニズムの結果としてASに至った例である。
臨床的マネジメント
症状に対する治療:
てんかん発作に対しては、抗痙攣薬を使用する。多動性行動、ならびに夜寝ないで家庭を壊すような行動に関しては、施設の利用で対応する。破壊的あるいは自傷的行動に対しては、行動療法(behavior modification)が有効な場合がある。理学療法、作業療法、ならびに、拡大代替コミュニケーション用器材(例えば、絵カードやコミュニケーションボード)や手話などの非言語的コミュニケーションに重点を置いた言語治療を行う。学校の場では、個別的で柔軟性をもった対応を行う。胃食道逆流症、摂食障害、便秘、斜視に対しては通常の管理を行う。脊柱側彎に対しては、胸腰椎コルセットの使用、ないしは外科的介入を行う。足首の亜脱臼や回内、アキレス腱の突っ張りに対しては、必要に応じ、装具の使用や外科手術を行う。
定期的追跡評価:
新たな発作の発生ないし発作の変化、発達の進行状況、行動の問題、可動性、運動能力、胃食道逆流症、便秘、摂食の問題に関するモニタリングを来院ごとに行う。年長の小児については、食欲の過剰に起因する肥満の評価を行う。脊柱側彎に関する臨床診査を年に1度、斜視があるようなら生後1歳未満の段階で眼科的診査を行う。眼科的診査は2歳の段階でも行い、その後は眼科医の判断に従った間隔でフォローする。脊柱側彎に関する臨床診査は年に1度行う。
避けるべき薬剤/環境:
過興奮性、多動性の行動を抑えるための鎮静剤が過剰投与にならないようにする。動きの異常が発作と見誤られた場合や、発作のコントロール後も脳波の異常が残るような場合などに、抗痙攣薬の過剰投与に陥りやすい。
遺伝カウンセリング
AS罹患者は、通常、孤発例(家系内で1人だけの罹患者)で、de novoの遺伝学的変化の結果としてASに至ったもので、その場合の再発リスクはごく低い。これより出現頻度の低いものとして、常染色体顕性遺伝となるインプリンティングパターン関連の遺伝学的変化、あるいは再発リスクに幅が生じる遺伝学的変化に起因してASに至るような例も存在する。したがって、再発リスク評価を信頼性のあるものにするためには、発端者の背後にある遺伝学的メカニズムを特定するとともに、両親の遺伝学的状態を確認しておくことが必要となる。現時点でASを引き起こすことがわかっている15q11.2-q13領域のすべての分子遺伝学的変化について、出生前の検出が可能である。したがって、背後にある遺伝学的メカニズムが判明している発端者については、出生前検査が1つの選択肢となる。
診断
アメリカAngelman症候群財団の学術諮問委員会との連携のもと、Angelman症候群(AS)の臨床診断基準がコンセンサスを得るに至っている[Williamsら2006]。ASの診断に関するレビューもいくつか存在する[Bird 2014,Buitingら2016,Prasadら2018]。
本疾患を示唆する所見
以下に示す臨床所見、検査所見、X線写真所見を有する例については、ASを疑う必要がある
臨床所見
- 出生前と出生時の経過に異常がみられず、出生時の頭囲は正常、そして特段の奇形もみられない。
- 生後6-12ヵ月の段階で発達指標の到達に遅延がみられ、最終的には重度に分類される発達遅滞に至るものの、機能の喪失には至らない。
- スピーチの障害がみられ、言葉はごくわずか、あるいは全く出ない。表出言語に比べると、受容言語や非言語コミュニケーションの能力は高い。
- 運動障害あるいは平衡障害、多くの場合、失調性歩行や四肢の振戦がみられる。
- よく笑いよく笑みを浮かべる、見るからに愉快そうな振る舞い、興奮しやすさ(しばしば手をばたつかせる動きをとる)、多動性行動といったものがさまざまな組み合わせで現れる行動の特異性がみられる。
診断の確定に資する臨床的基準[Williamsら2006]
罹患者の80%超にみられる所見
- 頭囲の成長の遅延、あるいは全身の成長に比べて遅い頭囲の成長
- てんかん発作
- 振幅の大きな遅棘波の特徴的パターンを伴う脳波の異常
2歳には絶対的小頭症もしくは相対的小頭症に至る。
これは3歳より前に始まることが多い。
罹患者の80%未満にみられる所見
- 平坦な後頭部、後頭部の溝、横幅の広い口、大きな歯間空隙、舌の前突、下顎前突といった頭蓋顔面症候(図1参照)
- 乳児期にみられる摂食の問題ないし筋緊張低下、舌突出癖、吸啜/嚥下障害、流涎、物を過度に長く口に入れて噛む行動
- 斜視
- 皮膚の低色素沈着、家系内の人と比べ薄い髪色や眼の色
- 下肢の深部腱反射の亢進
- 歩行中に特に多くみられる腕を曲げて持ち上げた姿勢
- 足首の回内あるいは外反を伴う歩隔の拡大
- 熱に対する感受性亢進
- 睡眠-覚醒サイクルの異常と睡眠要求度の低下
- 水への興味・関心、しわしわ感のある特定の紙やプラスチックへの興味
- 食物に関連する行動の異常
- 肥満(これは小児期後期にみられる;15q11.2-q13の欠失を有しない例のほうに多くみられる)
- 脊柱側彎
- 便秘
これらは15q11.2-q13の欠失を有する例のみにみられる。

図1:
ここに挙げた例はいずれも、分子遺伝学的にAngelman症候群(AS)の診断が下りたものである。
愉快げな表情、腕を上げた不安定な歩行が多く認められる。
顔貌からASが示唆されるようなことが時にあるものの、顔の症候はASに特異的なものではないことが多い。
検査所見
15q11.2-q13ゲノム領域の欠失(染色体マイクロアレイその他の方法で検出される)は、ASを示唆するものではあるが、それだけでASの診断が確定できるわけではない。
代謝、血液、化学的検査結果のプロフィールは正常である。
X線写真所見
脳の画像診断では、MRIでもCTでも、脳の構造に異常はみられないが、軽度の皮質萎縮や髄鞘形成異常がみられる場合もある。
診断の確定
発端者におけるASの臨床診断は、臨床診断基準(「本疾患を示唆する所見」の中の「診断の確定に資する臨床的基準」の項を参照)[Williamsら2006]をもとに確定させることができ、発端者における分子診断は、ASを示唆する所見を有することに加え、分子遺伝学的検査にて母親由来のUBE3Aアレルの発現や機能の障害を示唆する所見が得られることをもって確定させることができる(表1参照)。
分子診断
発端者におけるASの診断は、これを示唆する所見を有することに加え、分子遺伝学的検査で以下の1つが確認されることをもって確定する(表1参照)。
- 以下のもののうちの1つに起因する15q11.2-q13のメチル化異常
- 母性継承の15q11.2-q13領域(UBE3Aを含む領域)の欠失
- 父性染色体の15q11.2-q13領域の片親性ダイソミー(UPD)
- 母性染色体の15q11.2-q13領域のインプリンティング異常
- 母親由来のUBE3Aの病的バリアント
診断を確定させるための分子遺伝学的検査のアプローチは、ASを示唆する臨床所見あるいは検査所見をもとに行われることになる。
症候を有しつつも、まだ分子遺伝学的検査を受けたことがない例については、臨床所見に基づいて以下の形で進める。
- DNAメチル化解析
通常は、最初にDNAメチル化解析を行う。
15q11.2-q13領域の5-7Mbの欠失、UPD、インプリンティング異常に起因してASに至った例は、非メチル化(すなわち「父性発現」)状態(すなわち、異常な形の片親特異的DNAメチル化インプリンティング)のみを有している。
DNAメチル化解析により、AS罹患者の約80%が同定される。
注:商業ベースで利用可能なDNAメチル化解析では、15q11.2-q13の欠失、UPD、インプリンティング異常の3者の区別ができない。
背後にある分子メカニズムの特定には、別の検査を追加する必要がある(「遺伝カウンセリング」の項を参照)。
DNAメチル化解析で異常がみられなかった場合は、以下を行う。
- 単一遺伝子検査
最初に、遺伝子内の小欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントを検出する目的で、UBE3Aの配列解析を行う。
注:用いる配列解析の手法によっては、単一エクソン、複数エクソン、遺伝子全体といった単位の欠失/重複が検出されないことがある。
用いた配列解析の手法でバリアントが検出されなかった場合、次のステップとして行うべきものは、エクソン単位あるいは遺伝子全体の欠失や重複を調べるための遺伝子標的型欠失/重複解析である。
- マルチ遺伝子パネル
現況の表現型と直接関係のない遺伝子の意義不明バリアントや病的バリアントの検出を抑えつつ、疾患の遺伝的原因の特定に最もつながりやすいのは、UBE3A(例えば、てんかん、自閉症、知的障害用マルチ遺伝子パネルであれば、ほとんどのものがこれを含むデザインになっている)その他の関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネルである。
注:(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、今このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝学的検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
- 網羅的ゲノム検査
ASの症候を呈するにもかかわらず、DNAメチル化解析とUBE3Aの配列解析(ないしマルチ遺伝子パネル)で診断がつかなかった例については、利用可能なようであれば、エクソームシーケンシング、ゲノムシーケンシング、ミトコンドリアシーケンシングなどの網羅的ゲノム検査も視野に入ることになろう。
網羅的ゲノム検査の基礎的情報についてはここをクリック。
ゲノム検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
染色体マイクロアレイ(CMA)、蛍光in situハイブリダイゼーション法(FISH法)、核型解析*等で、15q11.2-q13の欠失が判明した例については、検査結果に基づいてDNAメチル化解析を行い、その欠失が母親由来の15番染色体に生じたものであることを確定させることになる。
*AS罹患者の1%未満は、細胞遺伝学的手法で視認可能な15q11.2-q13領域を含む15番染色体の再構成を有している。
表1:Angelman症候群で用いられる分子遺伝学的検査
| 方法 | 検出される遺伝学的メカニズム1 | 全発端者の中でその手法で検出可能なものの占める割合2 | ||||
|---|---|---|---|---|---|---|
| 15q11.2-q13欠失 | UPD | インプリンティング異常 | UBE3Aの配列バリアント | UBE3Aの欠失/重複 | ||
| DNAメチル化解析3,4 | X | X | X5 | 80%近く | ||
| メチル化特異的MLPA法6 | X | X | X | 80%近く | ||
| FISH法7 | X | 68%近く | ||||
| CMA8 | X | X9 | 70%-75%近く | |||
| UPD解析10 | X | 3%-7%近く | ||||
| ASインプリンティングセンター欠失解析11,12 | X | 0.3%未満 | ||||
| UBE3Aの配列解析13 | X | 11%近く | ||||
| UBE3Aの遺伝子標的型欠失/重複解析11,14 | X | 稀 | ||||
- 詳細は、「分子遺伝学」の項を参照。
- ASの臨床診断を受けている例の約10%については、本表にある検査結果がすべて「異常なし」と出る[Williamsら2010]。
- AS罹患者の中で、15q11.2-q13の5-7Mbの欠失、片親性ダイソミー(UPD)、インプリンティング異常に起因して生じた例については、非メチル化(すなわち「父性」)アレルのみ(すなわち、異常な片親特異的DNAメチル化インプリンティング)となっている。
- DNAメチル化解析では遺伝学的メカニズムの判別まではできない。
- インプリンティング異常の90%超は、母体の卵形成期あるいは胚発生初期に生じるエピゲノムの病的バリアントであると考えられている[Buitingら2016,Beygoら2019]。インプリンティング異常がインプリンティングセンターの欠失であるのか、それともエピゲノムの異常であるのかの判別作業は、主として研究機関の実験室でのみ可能である。
- Beygoら[2019]
- D15S10ないしSNRPNのプローブを用いたFISH解析により、ありふれた15q11.2-q13の欠失は検出可能であるが、通常、この欠失は、ふつうの細胞遺伝学的解析では検出することができない。
- 15q11.2-q13の欠失については、FISHよりCMAのほうがわずかに高感度で、欠失のサイズに関する情報もより詳しく得られる。CMAの場合は、ゲノムの他の領域に生じた欠失や重複を同時に検出することができる。
- SNPベースの染色体マイクロアレイによりUPDの検出は可能であるが、片親性ヘテロダイソミーの検出はできない。
- 多型DNAマーカーを用いて、UPDを検出することができる。 ただ、これには、罹患者と両親についてDNAサンプルの採取が必要である。
- 遺伝子標的型欠失/重複解析では、遺伝子内、もしくは他の標的領域の欠失や重複が検出される。具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失/重複の検出を目的に設計された遺伝子標的型マイクロアレイなど、さまざまなものがある。
- インプリンティングセンターの異常を示すのはAS罹患者全体の3%であるが、インプリンティングセンター内に検出可能な小欠失を有するのは、その10%未満である。
- 配列解析を行うことで、benign、likely benign、意義不明(VUS)、likely pathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、遺伝子内の小欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。UBE3Aはインプリンティングを受けた遺伝子であるため、父性継承の確認されたVUSについては、benignへと分類が下がる。配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
- CMAでは、通常、15q11.2-q13の大きな欠失が検出されるが、稀に、UBE3Aの複数エクソンの欠失や遺伝子全体の欠失が検出される場合がある。
ASの臨床診断を受けた例の約10%については、ASを引き起こす遺伝学的異常が検出されない。その理由として考えられるものには、以下のようなものがある。
- 臨床診断そのものが誤っていた可能性
- UBE3Aの調節領域に、検出されていない病的バリアントが存在する可能性
- UBE3Aの機能に関与する未知のメカニズムや遺伝子が存在する可能性
臨床的特徴
臨床像
Angelman症候群(AS)は、重度の発達遅滞と知的障害、重度の言語障害、失調性歩行ないし四肢の振戦、頻繁な笑い・ほほ笑み・興奮等の見るからに愉快そうな振る舞いを伴う独特の行動を特徴とする疾患である。小頭症とてんかん発作も多くみられる。発達遅滞が初めて確認されるのは生後6ヵ月前後であるが、ASの独特の臨床症候が顕在化するのは1歳を過ぎてからである。
表2:Angelman症候群:代表的症候の出現頻度
| 症候1 | その症候を有する例の割合 | コメント |
|---|---|---|
| てんかん発作 | 90% | 多くのタイプの発作あり;非痙攣性てんかん重積状態が20%にみられる。 |
| 非てんかん性ミオクローヌス | 20% | 成人により多くみられる。 |
| 睡眠の問題 | 80%-90% | 頻繁な覚醒,睡眠障害,不規則な睡眠-覚醒サイクル |
| 行動の症候 | 100% | 多動性,何でも口に入れてみる,頻繁なほほ笑みや笑い,攻撃性,不安,自閉症スペクトラムの形質 |
| 運動発達遅滞/振戦 | 100% | |
| 言語発達遅滞/高次脳機能障害 | 100% | インプリンティングセンターの異常をモザイクで有する例は、言語発達が比較的良好であることがわかっている。 |
| 摂食/消化器の問題 | 85% | |
| 小頭症1 | 25%-80% | 2歳までに明らかになる;15q11.2-q13欠失の例に最も多くみられる。 |
| 特徴的顔面症候 | 80%未満 | 平坦な後頭部,後頭部の溝,横幅の広い口,大きな歯間空隙,舌の前突,下顎前突;これに毛髪・皮膚・眼の低色素沈着が加わることあり。 |
| 斜視 | 40%-50% | |
| 脊柱側彎 | 50%以下 | 子ども:10%-30%;成人:30%-50% |
- 頭囲が正常であっても、それによりASの可能性が排除されることにはならない。
発作の発症時期
発作の発症は、1歳から3歳の間であることが多いものの、あらゆる年齢に分布する。
そして、大多数の例では5歳までに出現する。てんかんは、90%近くの例でみられ、特に15q11.2-q13欠失例に多くみられる[Khanら2019,Bindels-de Heusら2020]。発作は、多くの場合、全般的かつ特徴ある脳波の変化を伴う。具体的には、間欠的な棘徐波発射を伴う高振幅のデルタ活動のラン(時に切れ込みのあるデルタパターンとしてみられる)、広範囲に出現する律動性シータ活動のラン、小さな複数の棘波が複合波を形成する頭の後部3分の1における5-6Hzの律動性で鋭いシータ活動のランである。こうしたものは、閉瞼により誘発されることが多く、閉瞼時のみにみられるといったこともある[Boydら1988,Samanta 2021]。
発作のタイプは全くもってさまざまで、最も多くみられるものは、ミオクローヌス性発作、脱力発作、全般強直間代発作、非定型欠神発作である[Thibertら2009,Fiumaraら2010]。複数タイプの発作を示す例が50%近くに及ぶ。点頭てんかんは稀である。発作は成人期まで存在し続ける。
脳のMRIにて萎縮と軽度の髄鞘形成不全が明らかになることがあるものの、構造的病変はみられない[Hartingら2009,Castro-Gagoら2010]。
非痙攣性てんかん重積状態(NCSE)
NCSEは、子どもにおいても[Bindels-de Heusら2020]、成人においても[Prasadら2018]生じることがある。この状態は、臨床的には把握しにくいことが多いものの、機能や意識の低下を伴って、何時間も、時に何日間も続くことがある。最も多いのは非定型欠神発作型、あるいはミオクローヌス型NCSEで、ぼーっとした状態、非定型的欠神状態、脱力性の頭部落下、筋緊張低下、ミオクローヌス性運動等が出現する[Elia 2009,Wordenら2018]。
非てんかん性ミオクローヌス(NEM)
NEMは、皮質性ミオクローヌスとも呼ばれる。NEMと、特徴的脳波を呈する真性の発作とは、きちんと区別する必要がある。NEMにおいては、律動的でチック様、痙縮様の動きがみられるものの、意識には明らかな変化がみられず、てんかんでみられるような脳波の変化も伴わない。NEMがみられるのは、通常、ティーン世代と若い成人である[Pollackら2018]。
睡眠の問題
- AS罹患者においては睡眠の問題が多くみられる。具体的には、早くかつ頻回に目覚める、睡眠障害(入眠・睡眠維持困難)、切れ切れで不規則な睡眠-覚醒サイクル、一定時間笑い続けるといった夜間の異常行動、睡眠関連発作などがある[Pelcら2008,Spruytら2018]。
便秘、胃食道逆流症、脊柱側彎があると、睡眠障害にさらに拍車がかかることになる[Bindels-de Heusら2020]。睡眠関連の問題は年齢とともに改善することもあるが、一部には、いつまでも添い寝が必要な例もみられる[Walzら2005,Dosierら2017]。行動の問題、発作、睡眠の問題が併存することから、こうした問題への対処にあたっては、行動の改善、てんかんのコントロール、投薬、睡眠衛生の改善といったことに広く焦点を当てた治療とする必要がある。
行動の症候
行動面では、頻繁な笑いやほほ笑み、見るからに愉快そうな振る舞い、興奮してよく手をばたつかせる、多動性行動といった特徴がみられる。一部の乳児には、過度の笑いもしくは笑い発作を伴う、見るからに愉快そうな感情がみられる。乳幼児は、手やおもちゃを口に入れ続けたり、物をとっかえひっかえしたりといった絶え間なく見えるような活動を示すことがある。幼児期以降の探索行動としての遊びは、口で何かをもてあそんだり噛んだりといったことになっていく傾向がみられる。ASの幼児は基本的に全員、多動の要素を有している。そうした傾向は男女とも同じであるように思われる。おかしいと感じられる状況で笑いが出ることももちろんありはするが、それよりも、おかしくも何ともない出来事(心や体への刺激)に対する反応として、あるいは不安の表現として笑いが現れることのほうが多い。つまむ、握る、噛む、叩く、殴るといった攻撃的で自傷的な行動がみられることもある。こうした行動は、悪意というよりむしろ、他者の興味を引きたい思いとか、コミュニケーションの難しさに起因するフラストレーションの表れであることが多い[Arronら2011,Sadhwaniら2019]。AS罹患者は、ありとあらゆる種類の感情があり、家族や友だちと意味のある関係を築くことができ、家事・レクレーションその他の活動に参加することができる。
自閉症スペクトラム障害を示唆するような行動(例えば、水への興味や、しわしわ感のある紙やプラスチックへの興味、熱への過剰な感受性、食関連の行動の異常)がみられることがあるものの、社会との結びつきは概して良好である。おもちゃを並べる、回転するものや光るものに惹かれるといった定型的行動がみられることはほとんどない。AS罹患者の中には、ABA(応用行動分析)療法に良好な反応を示す例もみられる[Walz & Baranek 2006,Moss & Howlin 2009,Summers 2012]。
一般に、この行動プロフィールは成人期まで続いていく。ティーン世代から成人期にかけて特に問題となることとしては、欲求や嗜好を他者に伝える際のフラストレーション、感覚刺激や社会的関心を求める際のフラストレーション、嫌なことを避ける際のフラストレーションなどがある[Larsonら2015]。
運動発達と振戦
振戦は生後12ヵ月未満の段階からみられることがあり、これには深部腱反射が関係している(「臨床像」の中の「非てんかん性ミオクローヌス」の項を併せて参照)。
ASは、粗大運動発達指標の遅れと筋緊張低下がきっかけとなって幼児期に最初に疑われることが多い。ただ、軽症の子どもはかなり正常に近い歩き方をし、爪先歩きや跳ね歩きが少なく、時に前傾姿勢の歩き方をする。じっと立たされていると、不安や硬直が生じることがある。歩行開始年齢は、平均的には2.5歳から6歳の間である[Lossieら2001]。ASの子ども100人を対象とした最近の研究によると、15q11.2-q13欠失の例で歩行開始は平均で58ヵ月、欠失をもたない例は平均で41ヵ月であったという[Bindels-de Heusら2020]。重症の子どもの歩き方は、歩隔が広く、腕を上げて前腕を前屈させた、ぎくしゃくしたロボット様の硬い歩き方である。
随意運動は、不規則に見えるような動きとなることが多い。ごく軽症例の少しぎこちないといった程度のものから、重症例の調和に欠けた粗い動きまで、分布は連続的である。こうした動きのため、物を取ったり、食事をしたり、歩いたりといった作業ができなくなることがある。自立歩行が達成できない原因として、振戦、てんかん、視覚の問題、筋緊張異常、平衡感覚の問題等に起因する不安定性が関係しているように思われる。10%の子どもは歩行を達成できない[Clayton-Smith 1993]。
言語障害と認知機能の遅れ
言語や認知機能に関しては、重度の遅れがみられる。正式な精神測定検査では、発達の達成時期の遅れはおおむね24-30ヵ月の間にあるようであるが、言語障害や、多動・注意欠如といった行動の問題があるため、発達検査そのものが難しいという現実がある[Petersら2004]。認知機能については、検査で捉えられる値より実際のほうが高い可能性があるものの、それでも遅れは依然として重度と言えるレベルにあることが多い。15q11.2-q13の欠失を有する例の認知機能の遅れは、通常、あらゆる領域にわたって最重度である。
1,2語であっても、それが一貫して適切な使われ方をすることは稀である。赤ちゃんや幼児は、クーイングや喃語がふつうより少ない。18ヵ月になると、「ママ」のような単語が1つだけ出るようになることもあるが、見境なくこれが使用されることが多い。47人を対象とした調査では、39%が4語以内にとどまっていたとされているが、これらの語が目的をもって使用されていたがどうかは明らかにされていない[Buntinxら1995]。Larsonら[2015]は、13%が5語以上を有していたと報告している。受容言語の発達の進み方は、例外なく表出言語の発達の進み方を上回っていた[Bindels-de Heusら2020]。
発作や顕著な多動があると、アイコンタクトを含む初期のコミュニケーションの発達が阻害される可能性がある。それでも、ASの年長の子どもや成人については、指差しや直接触って指示する、ジェスチャーを使う、体の一部を指す、コミュニケーションボードを使うといった手段でコミュニケーションをとることが可能である。手話をスムーズに使いこなすことはできない[Larsonら2015,Pearsonら2019]。ただ、インプリンティングセンターの異常をモザイクで有する例の一部は、かなりの言語を有し、短い文を話し、最大60語が使用できる[Fairbrotherら2015,Le Fevreら2017]。
摂食と消化器の問題
乳児期初期は、吸啜障害のため母乳の哺乳や哺乳瓶での哺乳が難しい上に、筋緊張低下も有することがある。50%近くに哺乳不良がみられ、特に15q11.2-q13の欠失を有する例でそれが顕著にみられる。約10%-15%については、胃瘻や経鼻胃管が必要となる[Glassmanら2017,Khanら2019,Bindels-de Heusら2020]。
胃食道逆流症(GERD)がAS罹患者の45%-65%にみられ、これにより乳児では体重増加不良や嘔吐が生じる[Glassmanら2017,Khanら2019]。親が最初にこれに気づくきっかけとしては、嚥下困難や吐き出し、呼吸障害、喉詰まり、背中の反り返り、摂食拒否、痛み、摂食時の不快感などがある。GERDについては、上部消化管出血や食道炎を予防するための治療が必要となる。GERD関連の問題は、生涯続く可能性がある[Larsonら2015]。
嘔吐は珍しくなく、周期性であることもあれば間欠性であることもある。15q11.2-q13の欠失あるいは片親性ダイソミー(UPD)の例には、周期性の嘔吐がより多くみられるように思われる。嘔吐(病気や食物アレルギーに起因するものでないもの)の背景には、不安と行動の問題、薬剤の副作用、便秘などのさまざまな要因が関与していることが考えられる[Glassmanら2017]。
過食、ならびに食関連の問題行動は、すべての遺伝学的サブタイプでみられ、その発現頻度は20%-50%である[Welhamら2015,Bindels-de Heusら2020]。
便秘も多くみられ、これは年齢を問わずみられる。その症状としては、硬い便、排便頻度の減少、食欲の不振あるいは悪化、嘔吐、腹痛などがある。便秘は、行動の変化、体重減少、睡眠の質の低下、発作の増加といったものにつながる可能性があるため、適期に適切な対応をとることが重要である[Glassmanら2017,Khanら2019]。
小頭症
頭の成長の遅延あるいは相対的鈍化により、2歳までに、後頭部の平坦化を伴うことの多い絶対的小頭症あるいは相対的小頭症(-2SD未満)がしばしば生じる。報告されている小頭症の発現頻度には、25%から80%までばらつきがある。小頭症は、15q11.2-q13欠失の例により多くみられる。仮に頭囲が正常であったとしても、それによりAngelman症候群の可能性が除外されるわけではない[Tanら2011,Bindels-de Heusら2020]。
特徴的顔面症候
平坦な後頭部、後頭部の溝、横幅の広い口、大きな歯間空隙、舌の前突、下顎前突といったものが、罹患者の80%未満で報告されている(図1参照)。AS罹患者、中でも15q11.2-q13欠失例は、家系内の他の人より髪・皮膚・眼の色が薄いことが多い。舌は、形や大きさに問題はみられないものの、約30%-50%に持続性の舌突出がみられる。中には、この問題が成人期まで遷延する例もみられる。流涎は、皮膚のただれや誤嚥の要因になることはあっても、ふつうは重大な合併症に至ることはない。これに対する外科的治療や薬物治療(例えば、唾液腺管移動術や局所的なスコポラミンパッチの使用)は、通常、有効ではない。通常は、よだれ掛けや、時に作業療法といった保存的対応を行うことになる[Boyce & Bakheet 2005,Scullyら2009]。
斜視その他の眼の所見
斜視の発生頻度は40%-50%で、分子レベルの病因による違いはみられない[Tanら2011,Khanら2019,Bindels-de Heusら2020]。OCA2は、15q11.2-q13領域に座位する遺伝子で、皮膚・毛髪・光彩の色素沈着に一定の役割を有している。斜視は、眼の低色素を招来する遺伝性疾患に多くみられる傾向にある。網膜の色素は、視神経経路の正常な発生にとって決定的に重要である。AS罹患者においては、虹彩と脈絡膜(中心窩ではない)の低色素沈着が報告されているが、こうした低色素沈着は15q11.2-q13の欠失を有しない例でもみられることがあることから、OCA2だけで説明がつくものでもなさそうである。斜視に対しては、眼鏡やアイパッチ、そして適切と考えられる場合は手術も含めた標準治療が行われる。ただ、多動があることで治療への協力に問題が生じることも考えられる。斜視の手術が必要になるのは約30%であるが、手術では、おおむね良好な成績が得られている[Yeら2019]。
屈折異常の中で最も多くみられるのは乱視である。円錐角膜がみられることもあるが、この円錐角膜は、長期にわたり眼をこすったりえぐったりといった行動その他の原因で、二次的に生じたものである可能性がある。その他の眼の所見としては、近視、遠視、眼振、視神経萎縮、乳頭蒼白、網脈絡膜萎縮、眼瞼下垂、弱視などがある[Michielettoら2011]。
整形外科的問題
思春期に脊柱側彎が発症することがあり、年齢が上がるにつれてより多くみられるようになる[Giroudら2015,Larsonら2015]。側彎は、子どもの10-20%にみられる。成人では少なくとも30%-50%にみられ、通常は胸椎の側彎である。腰椎の過度の前彎が成人の20%-25%で報告されている[Sachdevaら2016,Prasadら2018]。側彎により可動性が制限されることがあり、進行予防のため装具での治療が行われる。重度の側彎を抱える例については、外科的改善が必要になる場合もある。これら以外の整形外科的合併症としては、股関節形成不全と骨密度低下などがある。骨減少症は、抗痙攣薬による治療が行われた例で、より高頻度にみられるようである[Coppolaら2007,Larsonら2015]。
思春期発達
AS罹患者の思春期発達は、ふつう正常である。Larsonら[2015]は、早発月経が17%、遅発月経が27%と報告している。思春期に生じるホルモンの変化が、行動・てんかんの両面に影響を及ぼす可能性がある。受胎能力は正常であると思われ、男女両性とも、子を儲けることは可能なようである。月経調節の選択肢を模索するため、婦人科医と面談することが望ましい[Kaskowitzら2016]。Lossie & Driscoll [1999]は、ASの母親から胎児への15q11.2-q13欠失の伝達例を報告している。
成長
出生時の身長、体重、頭囲は、ふつう正常である。AS罹患者の小児期の平均身長は一般集団の値を下回るものの、成人期の最終身長について言うと、大多数が正常の身長を獲得する。AS罹患者では、体重対身長比の増加がみられるが、これは、15q11.2-q13の欠失を有しない例により多く報告されている[Mertzら2014,Carsonら2019,Bindels-de Heusら2020]。
予後
若い成人の身体的健康状態は、概して良好であるように思われる。ただ、発作、異常な動き(運動失調,歩行機能の低下)、行動の問題に加え、すでに述べた消化器、睡眠、整形外科的問題等については、成人期を通じて継続する場合がある。ASの成人のその他の健康上の問題に関しては、一般集団と同程度のリスクであるように思われる。ASの成人については、自立した生活を営むことは不可能である。多くの人は自宅、もしくは自宅に似た施設で生活を送っている。寿命に関するデータは得られていないが、寿命は正常に近いように思われる[Larsonら2015]。
遺伝型-表現型相関
ASは、分子レベルの原因が何であっても、すべて、重度から極度の知的障害、運動障害、特徴的行動、重度の言語障害から成る同様の表現型となって現れる。ただ、遺伝型によって多少の表現型の違いが現れることはある[Tanら2011,Valenteら2013,Bindels-de Heusら2020,Keuteら2020]。大まかに言うと、次のような相関である。
- 15q11.2-q13の5-7Mbにわたる欠失は、小頭症、てんかん発作、運動障害(例えば、運動失調,筋緊張低下,摂食障害)、言語障害といったものを伴う最重度の表現型の形で現れる。一方、BMIについては、欠失例のほうがUPDやインプリンティング異常の例より低めとなる。欠失のサイズが大きなもの(例えば、BP1-BP3の切断[クラスⅠ;ISCA-37404])と小さなもの(例えば、BP2-BP3の切断[クラスⅡ;ISCA-37478])とで臨床症候の違いがみられるかどうかという点に関しては、よくわかっていない(図2参照)。
- UBE3Aの病的バリアントを有する例やインプリンティング異常の例は、UPDの例より臨床症候が軽度のようである。
- UBE3Aのトランケーションバリアントは、UBE3Aのミスセンスバリアントよりも臨床症候が重度になりやすい[Keuteら2020]。
- 欠失以外の理由で生じたインプリンティング異常をモザイクで有する例(インプリンティング異常例の20%近くを占める)は、最も高い言語能力を有しており[Nazlicanら2004]、最大50-60語を話し、簡単なものながら文の使用も可能である[Fairbrotherら2015,Le Fevreら2017]。
- OCA2を含んだ15q11.2-q13の欠失を有する例は、虹彩、皮膚、毛髪が低色素沈着であることが多い。
OCA2は、皮膚、毛髪、虹彩の色素の発達に関連するチロシンの代謝に重要なタンパク質をコードしている(「眼皮膚白皮症2型」のGeneReviewを参照)。
ただ、UBE3Aも体細胞組織中のメラノコルチン1受容体(MCR1)活性を調節することが知られており、そうしたことから、AS罹患者の相対的低色素沈着に関しては、OCA2以外にも複数の要因が関与している模様である[Low &Chen 2011]。
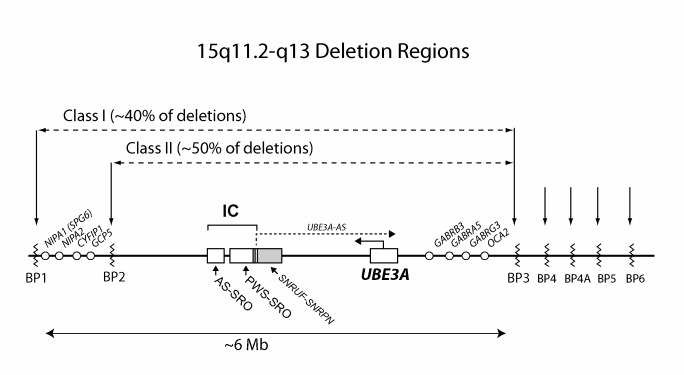
図2:切断点領域BP1-BP6を表す15q11.2-q13領域の模式図
これらの切断点領域には低コピーリピートのエレメントが存在する(詳しくは本文を参照)。
Angelman症候群を生じさせることになる染色体欠失の約90%はBP1あるいはBP2に始まり、BP3に終わるものである(それぞれ、クラスⅠとクラスⅡ)。約10%は、それより大きな欠失であるが、通常はBP1からBP5までであり、BP5を超える欠失は稀である。インプリンティングがなされておらず、二親性の発現を示す遺伝子は白抜きの○印で示した。2つの重要なインプリンティングセンター(IC)エレメントであるAS-SROとPWS-SROは白抜きの□印で示した。グレーの□で示したSNURF-SNRPN遺伝子は、PWS-SROと一部が重なっている。SNURF-SNRPNセンス/UBE3Aアンチセンス転写産物は、UBE3A-ASと表示している。
浸透率
UBE3Aの病的バリアント、インプリンティングセンターの欠失、UBE3Aを含む15q11.2-q13の微小欠失[Kurodaら2014]、父性アレルに影響を及ぼすある種の染色体転座については、不浸透の可能性がある(図3参照)。
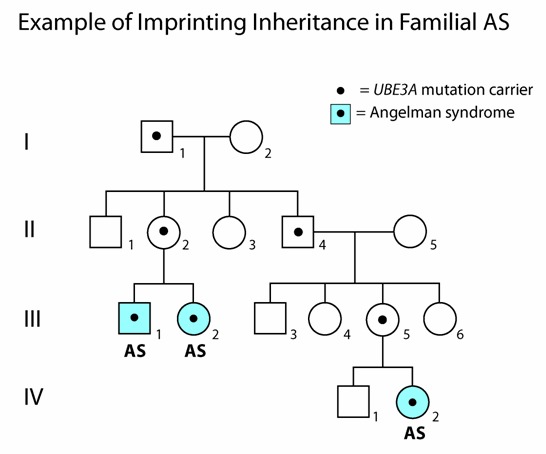
図3:Angelman症候群(AS)におけるインプリンティングの継承を説明する系図
男性(最上段左,Ⅰ-1)からUBE3Aの病的バリアントを継承した2人の子ども(Ⅱ-2,Ⅱ-4)に影響は現れない。それは、変異したUBE3Aはすでに父親の生殖細胞内で不活化(すなわち、インプリンティングによる不活化)がなされているからであり、また、2人の子どもはともに母親(Ⅰ-2)から不活化されていない正常なUBE3Aを継承しているからである(正常な脳機能の上では、不活化されていないUBE3Aアレルが1つだけあればよい)。ところが、娘(Ⅱ-2)がUBE3Aの病的バリアントを男性の孫(Ⅲ-1)、女性の孫(Ⅲ-2)に伝達すると、この2人の孫はともにASとなる。それは、この2人が父親から継承したUBE3Aは不活化されているため、2人ともUBE3Aアレルが発現しないことになるからである。この2人の叔父の女性の孫(最下段,Ⅳ-2)にASが発生した理由も、これと同じである。
発生頻度
集団内でのASの発生頻度は、12,000人に1人から24,000人に1人と推定されている[Mertzら2013]。
遺伝学的に関連のある疾患
Prader-Willi症候群(PWS)
PWSは、本来、父性発現である15q11.2-q13領域のインプリンティング遺伝子の発現がみられないことに起因して生じる疾患である。PWSとAngelman症候群(AS)は、年長の子どもでは臨床症候にはっきりした違いがみられるものの、2歳未満の子どもでは臨床症候に一部重なりがみられる(例えば、摂食障害,筋緊張低下,発達遅滞)。
母親由来染色体の15q11.2-q13の中間部重複
これは臨床的に、ASともPWSとも明確に異なる障害を引き起こす。dup15q11.2-q13罹患者には顔面の形態異常はみられない一方で、軽度から中等度の学習障害があり、自閉症スペクトラムでみられる行動を有する例がある。「15q重複症候群とその関連疾患」のGeneReview(訳注:現在は、タイトルが「母性15q重複症候群(Maternal 15q Duplication Syndrome)」に変わっている)を参照されたい。
鑑別診断
Angelman症候群(AS)の乳児は、共通して、非特異的な精神運動発達遅滞ないしてんかん発作を呈する。したがって、鑑別診断の範囲も広く非特異的なものとなり、脳性麻痺、非活動性脳症、ミトコンドリア脳症といったものを包含するものとなる。AS乳児の大多数にみられる振戦やぎくしゃくした四肢の動きが、ASと他の疾患を鑑別するポイントになる場合がある。
ASに症候が類似した疾患に関するレビューが存在する[Tanら2014]。そこに挙げられた疾患、ならびに、鑑別を要するその他の疾患を表3に挙げた。
表3:Angelman症候群との鑑別診断に際して注目すべき遺伝子
| 遺伝子 | 疾患名 | 遺伝形式 | 特徴的症候 | 鑑別のポイントとなる症候 |
|---|---|---|---|---|
| ADSL | アデニロコハク酸リアーゼ欠損症(ADSLD,OMIM 103050) | AR | 精神運動障害,自閉症の症候,筋緊張低下,てんかん発作,運動失調,重度の言語障害,過剰な笑い,きわめて愉快な気質,多動,集中力低下,物を口に入れる性癖,癇癪,常同運動 | ADSLDでは:脳のMRIで脳萎縮ないし小脳萎縮がみられる場合あり。 |
| ATRX | X連鎖αサラセミア知的障害症候群(ATR-X) | XL | 小頭症,筋緊張低下,流涎,胃食道逆流症,愛想のよい行動 | ATR-Xでは:性器異常と骨格異常 |
| EHMT1 | Kleefstra症候群(KS) | AD | 重度の言語発達遅滞と小児期の筋緊張低下を伴う中等度から重度の知的障害 | KSでは:独特の顔面症候(眉毛癒合,下赤唇反転)と言語能力(軽症例では100語超の語彙があり、文が使えることあり) |
| HERC2 | HERC2知的障害(OMIM 615516) | AR | 発達遅滞,知的障害,筋緊張低下,独立歩行の遅延(2.5-5歳),走るとき腕を上げ肘を曲げ歩隔が拡大する。 | HERC2知的障害では:ちょっとしたことで笑うという性質がみられず、(一部の例では)比較的知的障害が軽度。 |
| MBD5 | MBD5ハプロ不全症 | AD | 発達遅滞,知的障害,てんかん発作,睡眠障害,異常行動(例えば、自閉症様行動,自傷,攻撃性) | MBD5ハプロ不全症では、振戦や興奮しやすく愉快げな行動はみられない場合あり。 |
| MECP2 | Rett症候群 | XL | 女性では、意図をもった獲得性の手の動き、ならびに音声言語あるいは言語能力(例えば、喃語)の部分/完全欠如;歩行の異常;手の常同運動(例えば、手を捩る/絞る,手を叩く/手で叩く,物を口に入れる,物を洗うような/絞るような機械的動作) | Rett症候群では:神経発達が退行性経過をとり、目的をもった手の使用がなく、(通常は)愉快そうな振る舞いもみられない。 |
| MECP2重複症候群 | XL | 男性では1、スピーチの発達がゼロもしくは極少の、重度から極度の知的障害、運動発達の重度遅延を伴う早期発症型の筋緊張低下、てんかん発作 | MECP2重複症候群では:下肢を中心とした進行性の痙性、反復性呼吸器感染症の形で現れる感染素因 | |
| MTHFR | 重症メチレンテトラヒドロ葉酸還元酵素欠乏症(MTHFR,OMIM 236250) | AR | 愉快そうな振る舞い、失調性歩行、スピーチの欠如、後頭部の平坦化を伴う1男児が報告されている2。 | MTHFRのほうが、筋緊張低下と関節弛緩がより重度であることが多い。 |
| SLC9A6 | Christianson症候群(CS) | XL | 男性では:発達遅滞/知的障害(通常は重度から極度);言語発達はみられないか極少;多動;てんかん(発症年齢は通常3歳未満);体幹動揺;出生後発症型の小頭症 | CSでは:進行性小脳萎縮(ふつうは10歳以降)と、生涯にわたる体重増加不良と低BMIの問題 |
| TCF4 | Pitt-Hopkins症候群(PTHS) | AD | 発達遅滞,知的障害,行動の異常(愉快げな気質と評されることが多い);大多数はスピーチの発達がみられないが、表出言語に比べ受容言語に相対的強みをもつ。 | PTHSでは、特徴的顔貌3,呼吸パターンの異常 |
| WAC | WAC関連知的障害(WAC-ID) | AD | 発達遅滞,知的障害,乳児期における筋緊張低下±口腔の筋緊張低下,新生児期の摂食障害,胃食道逆流症ないし便秘,行動の異常,呼吸の問題,反復性感染症,喘息ないし呼吸パターンの異常,視覚の異常 | WAC-IDでは、通常、知的障害の程度は軽めで、単語や文章を話す能力があり、てんかん発作の発症頻度が低めで、小頭症はみられない。 |
| ZEB2 | Mowat-Wilson症候群(MWS) | AD | 発達遅滞,知的障害,スピーチはみられないか極少ながら、受容言語は比較的残る;大多数に愉快げな振る舞いと歩隔の拡大がみられる。 | MWSでは、特徴的顔貌4と多発性先天奇形 |
| 40超の遺伝子 | 先天性N結合型グリコシル化異常症(CDG-N-linked) | AR (XL) |
稀ながら、罹患児が不安定性歩行、言語障害、てんかん発作を有するような場合は、ASに似ることがある。 | CDG-N-linkedは通常、乳児期にみられ、複数の器官系に臨床症候(例えば、成長障害,発達遅滞,肝障害,筋緊張低下/神経学的異常)が現れる。 |
AD=常染色体顕性;AR=常染色体潜性;XL=X連鎖性
- 女性でMECP2重複症候群が生じることは稀である。それは、重複断片をもつほうのX染色体が選択的に不活化されるからである。ただ、稀には、女性でも男性と同様の重症度をもつ例がみられ、男性の場合に似た臨床症候が現れることがある。
- Arnら[1998]
- PTHSの診断を行う上で、頭蓋顔面症候は重要であるが、乳児期にはそれほど目立たないことがある。発達に懸念のある乳児の中でPTHSを見つける上での最初の鍵は、多くの場合、鼻と下顔面の突出である。
- 眼間開離、内側が広がった太い眉、低くぶら下がった鼻柱、目立つもしくは尖ったオトガイ、口の開いた表情、中央部が凹んでもち上がった耳垂。
染色体異常
Phelan-McDermid症候群(22q13.3欠失)、Koolen-de Vries症候群(17q21.31欠失あるいはKANSL1のヘテロ接合性病的バリアントに起因して生じる)、1q21.1反復性微小欠失をはじめとするいくつかの染色体異常でも、AS類似の症候が現れることがある。これらの疾患では、非醜形性の顔面症候、言語と知能の障害、そして(一部の罹患者では)愉快げで興奮しやすい行動を特徴とする。
注:摂食障害と筋緊張低下を有するASの乳児については、染色体マイクロアレイやFISHで検出された15q11.2-q13の欠失が、DNAメチル化
解析で母親起源のものであるということが明らかになるまでの間は、Prader-Willi症候群と誤診される可能性がある(「遺伝学的に関連のある疾患」の項を参照)。
臨床的マネジメント
今のところ、Angelman症候群(AS)に関する臨床的管理のガイドラインとして公表されたものはない。最初の診断時に続いて行う評価
ASと診断された罹患者については、疾患の範囲やニーズを把握するため、診断に至る過程ですでに実施済でなければ、表4にまとめた評価を行うことが推奨される。
表4:Angelman症候群罹患者の最初の診断後に行うことが推奨される評価
| 系/懸念事項 | 評価 | コメント |
|---|---|---|
| 神経 | 神経学的評価 | 脳のMRI、脳波を含むものとする。 |
| 発達と行動 | 発達評価と行動評価 | 以下を含むものとする。
|
| 消化器/摂食 | 消化器/栄養/摂食チームによる評価 | 胃食道逆流症と栄養状態に関する評価を含むものとする。 |
| 眼 | 眼科的診査 | 斜視,眼白皮症の有無,視力に関する評価 |
| 筋骨格 | 整形外科/物理療法面,ならびにリハビリテーション/理学療法/作業療法面での評価 | 以下の評価を含むものとする。
|
| 遺伝カウンセリング | 遺伝の専門医療職1の手で行う。 | 医学的、個人的な意思決定の用に資するべく、本人や家族に対し、ASの本質、遺伝形式、そのもつ意味についての情報提供を行う。 |
| 家族への支援/情報資源 | 以下の必要性に関する評価
|
- 臨床遺伝医、認定遺伝カウンセラー、認定上級遺伝看護師をいう。
症候に対する治療
表5:Angelman症候群罹患者の症候に対する治療
| 治療 | 治療 | 考慮事項/その他 |
|---|---|---|
| 多動的行動 | 経験豊富な神経内科医による抗痙攣薬を用いた標準治療:
|
|
| 多動的行動 |
|
|
| 睡眠障害 | 周りをかき乱す夜間の覚醒を受け入れられる安全な監禁を実現した寝室 | 入眠の1時間前にメラトニン0.3mgを投与することが有益なことがある。 ただ、子どもが起きていても、夜中に使用すべきではない。 |
| 社会規律を乱す行動や自傷的行動 | 行動修正療法が有効な場合あり。 | |
| 運動発達遅 |
|
差別のない教室運営のためには、教師の補助役やアシスタントの配置とともに、特別な物的資源の供給が必要になる場合あり。 |
| 言語発達遅滞 |
|
個別的で柔軟な対応ができる学校にすることが、教育戦略として重要である。 |
| 胃食道逆流症 | 消化器医による標準治療:
|
噴門形成術が必要になる場合あり。 |
| 体重増加不良/成長障害 | 吸啜の弱さやぎこちなさに対処するため、新生児には摂食治療、特殊乳首その他の戦略が必要になる場合あり。 | |
| 便秘 | 高繊維剤や潤滑剤の使用がしばしば必要となる。 | |
| 視覚の異常ないし斜視 | 眼科医の推奨に従った標準治療 | 早期介入サービスあるいは学区を通じた地域の視覚サービス |
| 脊柱側彎 |
|
年長の成人は運動や活動が低下するため、活動をスケジュールに組み込むことが側彎と肥満の減少に有効な場合あり。 |
| 他の整形外科的症候 | 足首の亜脱臼あるいは回内、アキレス腱の突っ張りは、補装具や外科手術で改善できる可能性あり。 |
- Thibertら[2012]
- 治療の初期で広く用いられる抗痙攣薬としては、クロバザム、レベチラセタム、ラモトリギン、クロナゼパムなどがあり、これらについては臨床使用の調査報告が存在する[Shaayaら2016,Prasadら2018]。その他の発作薬が使用されることは比較的少なく、有効性に関するデータも十分には揃っていない(例えば、ブリバラセタム,セノバメート,フェルバメート,ガバペンチン,サコサミド,プレガバリン,ルフィナミド,チアガビン,カンナビジオール)。親からの聞き取り調査によると、フェノバルビタール、プリミドン、カルマバゼピン、フェニトイン、バルプロ酸、ビガバトリンは比較的効果が薄いようである[Noltら2003]。少数ながら、AS罹患者の中には、発作の頻度が低いため抗痙攣薬を要しない例もみられる。
- 非痙攣性てんかん重積の外来治療にはジアゼパムが有用な場合がある[Wordenら2018]。
- 非てんかん性ミオクローヌスの治療は難しいことが多いが、これまでにレベチラセタム、クロバザム、クロナゼパムが使用されている[Pollackら2018]。ぺランパネルが奏功したとの最近の報告がみられる[Kawanoら2020]。
- 発作歴のあるAS罹患者については、発作の遷延に対する救急薬(直腸ジアゼパムゲルやミダゾラム・ジアゼパムの鼻腔内投与)が使用できるようにすべきである[Fedak Romanowskiら2021]。両親/介護者に対し、てんかん発作の一般的な現れ方に関し、教育しておくことが望ましい。てんかんと診断された子どもに対する非医療的介入と対処法に関する情報については、「Epilepsy Foundation Toolbox」を参照されたい。
定期的追跡評価
表6:Angelman症候群罹患者で推奨される定期的追跡評価
| 系/懸念事項 | 評価 | 実施頻度 |
|---|---|---|
| 神経 |
|
来院ごと。 |
| 発達 |
|
|
| 精神/行動 | 多動的/自傷的行動、睡眠障害に関する行動評価 | |
| 筋骨格 | 運動能力、自助能力に関する物理療法、作業療法/理学療法的評価 | |
| 消化器 | 胃食道逆流症と便秘に関するモニタリング | |
| 栄養/摂食 |
|
|
| 眼 | 眼科的診査 | 斜視がある場合は1歳未満で;2歳までに一般的評価;眼科医の指定に従いフォローアップ |
| 脊柱側彎 | 臨床的診査 | 年に1度 |
避けるべき薬剤/環境
過剰治療
- AS罹患児は、運動の異常が発作と誤って捉えられること、ならびに発作のコントロール後も脳波の異常が残ることから、薬の過剰投与に至るリスクを有している。
- AS罹患者の行動上の表現型としては、過剰な興奮性、多動的行動、社会的コミュニケーション障害などがある。こうした問題のため、社会との軋轢を生じることに関し罹患者は高リスクとなる。時には、リスペリドン(リスパダール®)その他の非定型抗精神病薬を使用することで、限定的ながらもある程度の効果が得られることがある。こうした薬剤が必要な場合でも、過鎮静その他の副作用を避けるべく注意が必要である。
リスクを有する血縁者の評価
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
研究段階の治療
DNAメチル化の経路を増やして、中枢神経系におけるUBE3Aの父性アレルの発現を増加させるための試みとして、葉酸、ビタミンB12、クレアチン、ベタインを経口投与する臨床試験が行われてきたものの、最初の試験では有意の臨床的効果は明らかにならなかった[Petersら2010]。高選択性のGABA受容体作用薬である経口ガボキサドールを用いた現在進行中の研究でも、今のところ明らかな効果は確認されていない[Birdら2021]。経口レボドパ/カルビドパを用いた研究でも、ASへの有意の効果は明らかになっていない[Tanら2018]。現在、アンチセンス鎖のオリゴヌクレオチド媒介性のUBE3A発現増強を評価する2つの臨床試験が進行中である。1つは、Hoffman-La Roche社がスポンサーとなっており(molecule RO7248824)、もう1つはGeneTX Biotherapeutics社がスポンサーとなっている(molecule GTX-102; NCT0459281)。
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「Clinical Trials.gov」、ならびにヨーロッパの「EU Clinical Trials Register」を参照されたい。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
Angelman症候群(AS)罹患者は、通常、孤発例(家系内で1例のみの発生例)として現れ、de novoの遺伝学的変化に起因して疾患に至ったものであり、再発リスクはきわめて低い(例えば、母性継承の15q11.2-q13領域の欠失)。これより出現頻度は低いながら、常染色体顕性遺伝となるインプリンティングパターン関連の遺伝学的変化(例えば、UBE3Aの病的バリアント)、あるいは再発リスクに幅が生じる遺伝学的変化(例えば、不均衡型染色体転座)に起因してASに至るような例も存在する。したがって、再発リスク評価を信頼性のあるものにするためには、発端者の背後にある遺伝学的メカニズムを特定するとともに、両親の遺伝学的状態を確認しておくことが必要となる。
家族構成員のリスク
発端者の両親
発端者の親は非罹患者である。
発端者の同胞
-
AS罹患者の同胞の有するリスクは、発端者におけるASの遺伝学的メカニズム、ならびに、両親の遺伝学的状態によって変わってくる。
- 発端者がUBE3Aの病的バリアントを有しておらず、かつ、DNAメチル化パターンが母性発現の喪失を示すものであった場合は、遺伝カウンセリングを目的として、背景にある遺伝学的メカニズム(すなわち、母親由来の15q11.2-q13領域の欠失,父性の15q11.2-q13の片親性ダイソミー[UPD],母親由来の15q11.2-q13領域のインプリンティング異常)の特定が必要となる。
発端者に対して行うことが推奨される検査は以下の通りである。
- 最初に、15q11.2-q13欠失解析(染色体マイクロアレイもしくはその他の方法で)を行う必要がある。
- 15q11.2-q13の欠失が検出されず、かつ、マイクロアレイ(あるいは、コピー数バリアントの検出を目的に用いられたその他の方法)で分節型あるいは染色体全体のアイソダイソミーが確認されなかった場合は、染色体全体のヘテロダイソミーの可能性を排除する目的で、15番染色体のDNA多型解析を用いることになろう(こうしたヘテロダイソミーは、Prader-Willi症候群では比較的多くみられるものの、著者らの知る限り、ASでは報告されていない[Fridman & Koiffmann 2000])。
- UPDが検出されなければ、インプリンティング異常が存在するものとの推定が成り立つことになる。その場合は、インプリンティングセンターの欠失があるか否かを確定するための追加の検査を行うことになろう。
- 発端者の背景にある遺伝学的メカニズムが確定されれば、両親の遺伝学的状態を評価することが可能となる。
- 両親に対して行うことが推奨される検査(発端者の有する遺伝学的メカニズムをもとに推奨される検査)、ならびにそれに対応する同胞の再発リスクを表7にまとめて示した。
表6:Angelman症候群の遺伝学的メカニズムと親の遺伝学的状態別にみた発端者の同胞の有するリスク
| 分子的クラス1 | 家系の割合 | 発端者の遺伝学的メカニズム | 推奨される出生前検査 | 同胞のリスク |
| Ⅰa | 65%-75% | 15q11.2-q13.1の5-7Mbの欠失 | 母親:染色体・FISH解析 | 母親の染色体・FISH解析で異常なしなら1%未満2 |
| Ⅰb | 1%未満 | 不均衡型染色体転座あるいは15q11.2-q13.1領域の中間部小欠失の継承 | 母親:染色体・FISH解析 |
|
| Ⅱa | 3%-7% | 父性片親性ダイソミー(核型は正常) | 母親と父親:染色体分析 |
|
| Ⅱb | 1%未満 | 父性片親性ダイソミー(父性転座の素因) | ||
| Ⅲa | 0.3% | インプリンティングセンターの欠失を伴うインプリンティング異常 | 母親:インプリンティングセンターの欠失に関する標的型検査(表現型の上で異常のない母親の父親由来15番染色体にde novoのインプリンティングセンターの欠失があったり、父親から継承したインプリンティングセンターの欠失がある可能性あり6) |
|
| Ⅲb | 2.5%-3% | インプリンティングセンターの欠失を伴わないインプリンティング異常 | 1%未満(インプリンティングセンターの欠失を伴わないインプリンティング異常の発端者の家系におけるASの再発例は、現時点で報告されていない)7 | |
| Ⅳ | 11% | UBE3Aの病的バリアント | 母親:UBE3Aの病的バリアントに関する標的型検査 |
|
| Ⅴ | 10% | 「その他」-分子的異常が検出されず | 検査対象外 | リスクの判定は不能 |
- Jiangら[1999]による分類に準拠。
- 15q11.2-q13の欠失を母親が生殖細胞系列モザイクで有していた例の報告がみられる[Sánchezら2014,Tangら2019]。
- Torisuら[2004],Kurodaら[2014]
- ここに挙げたリスクの数字は、染色体に異常のないASのUPDの例に再発がみられた例がないこと、他の疾患におけるUPDの実態、UPDの発生メカニズムに関する理論的考察をもとに算定したもの。
- Harpeyら[1998],Bramswigら[2018]
- 理論的には、母親がインプリンティングセンターの欠失を生殖細胞系列モザイクで有している可能性が考えられるものの、著者らの知る限り、実際の報告例は存在しない。
- 2つのインプリンティングセンターエレメントを分離する1-1.5Mbの逆位を有するものの、インプリンティングセンターの欠失は有しない2人の同胞例に関する1報告がみられる[Buitingら2001,Williamsら2010]。
- 母親がUBE3Aの病的バリアントを体細胞/生殖細胞系列モザイクで有していた例の報告がみられる[Hosokiら2005]。
発端者の子
現在のところ、AS罹患者が子を儲けたという報告は1例にとどまっている[Lossie & Driscoll 1999]。子の有するリスクについては、正式な遺伝カウンセリングの場で判定することが必要である。
他の家族構成員
- 発端者の母親(片親性ダイソミーとRobertson転座の場合は父親)にUBE3Aの病的バリアント、インプリンティングセンターの欠失、染色体の構造的再構成が同定された場合は、ASの素因となるその遺伝学的変化を有していたほうの片親の同胞に対して、遺伝カウンセリングと遺伝学的検査の選択肢を提供することが必要である。
- 同定されたインプリンティングセンターの欠失あるいはUBE3Aの病的バリアントを、発端者の母親がヘテロで有していた場合、母親の同胞も同様に、インプリンティングセンターの欠失やUBE3Aの病的バリアントに関しリスクを有することになる。
非罹患者ではあるがヘテロ接合者である発端者の母方の伯父(叔父)について言うと、罹患者である子をもつリスクは有しないものの、罹患者である孫をもつリスクは有することになる。それは、その母方の伯父(叔父)の非罹患者である娘が、インプリンティングセンターの欠失あるいはUBE3Aの病的バリアントを継承している可能性があるからである。
関連する遺伝カウンセリング上の諸事項
家族計画
- 遺伝学的リスクの確定、出生前/着床前遺伝学的検査を受けるかどうかの話し合いに最も適しているのは、妊娠前の時期である。
- ASの子をもつリスクを有する若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法であるとか、遺伝子・病原メカニズム・疾患等に対するわれわれの理解が、将来はより進歩していくことが予想される。そのため、分子診断の確定していない(すなわち、原因となった病原メカニズムが未解明の)発端者のDNAについては、保存しておくことを検討すべきである。
出生前検査ならびに着床前遺伝学的検査
高リスクの妊娠に備えた出生前検査や着床前遺伝学的検査を行うには、発端者の背景にある遺伝学的メカニズムを事前に同定しておくことが必要である。
高リスクの妊娠
ASを引き起こす15q11.2-q13領域における既知の分子遺伝学的変化(すなわち、分子クラスⅠa,Ⅰb,Ⅱa,Ⅱb,Ⅲa,Ⅲb,Ⅳ;表7参照)については、絨毛膜絨毛サンプリング(CVS)あるいは羊水穿刺によって得た胎児細胞を用いてDNA解析ないし染色体/FISH解析を行うことで、すべて出生前に検出可能である。
CVSで得られた胎児細胞を用いてDNAメチル化解析(15q11.2-q13の5-7Mbの欠失、UPD、インプリンティングセンターの異常を有する例に対するDNAメチル化解析)を行うことは、理論上、可能である。ただ、DNAメチル化解析を用いた出生前検査を施行している少数の検査機関では、羊水細胞の使用を推奨している。それは、胎盤由来の細胞はメチル化の程度が比較的低いためである。CVSで得られた胎児細胞を用いてFISH解析、インプリンティングセンター欠失解析、UBE3Aの配列解析を行うことは、技術的には可能なはずである[Beygoら2019]。
出生前検査は、発端者の有する遺伝学的メカニズムが確定し、なおかつ、両親に対して再発リスクに関するカウンセリングを行った後で行うようにすべきである。それは、発端者の有する分子的原因の内容次第で、リスクの大きさも、用いる遺伝学的検査の内容も大きく変わってくるからである(「診断の確定」の項を参照)。
- 15q11.2-q13の欠失、あるいはUPDに起因してASの子どもを1人もつに至った両親で、両者とも染色体異常を有しない場合、再発リスクは低いものと思われるが、確認のための出生前検査を行うことも1つの選択肢である。
- UBE3Aの病的バリアントに起因してASの子どもを1人もつに至った両親については、仮に母親からUBE3Aの病的バリアントが検出されていなかったとしても、出生前検査を行うようにすべきである。それは、母親の生殖細胞系列モザイクの可能性が残るからである。
- 15番染色体の絡む転座については、再発リスクが高いため、転座の継承状態を調べる出生前検査が意味をもつことになる。
15番染色体の絡む転座の継承がみられた場合は、FISH解析と、どちらの親起源かを調べる検査(DNAメチル化解析ないし多型解析)を検討する必要がある。
低リスクの妊娠
ASの家族歴のない低リスクの妊娠の場合は、以下のような状況にあるときASを考慮する必要がある。
- 15q11.2-q13の欠失
CVSあるいは羊水穿刺で採取した試料の細胞遺伝学的解析から15q11.2-q13の欠失が疑われた場合は、欠失の確認を目的としたFISH解析あるいは染色体マイクロアレイ解析(CMA)が適応となる。15q11.2-q13の欠失が確定したら、その欠失がどちらの親起源かを調べる検査を行って、母親由来(その場合、胎児はAS)であるか、父親由来(その場合、胎児はPrader-Willi症候群[PWS])であるかを確認することとなる。
- 15トリソミーあるいは15トリソミーモザイク
CVSで15トリソミーあるいは15トリソミーモザイクが疑われた場合で、なおかつそれに続く羊水穿刺で染色体数が46本であることがわかった場合は、トリソミーレスキューによって片親の1本の15番染色体が失われてAS(父性UPD)あるいはPWS(母性UPD)に至っている可能性を検討することが絶対に必要である。こうした場合は、羊水細胞を用いてどちらの親起源かを調べるDNA検査を行うことになろう。
- 15番染色体の絡むde novoの転座あるいは15番過剰マーカー染色体
15番染色体の絡むde novoの転座あるいは15番過剰マーカー染色体が検出された場合は、15q11.2-q13の欠失(欠失のサイズはさまざま)あるいはUPDの可能性を評価するため、FISH解析もしくはCMAに加えて、どちらの親起源かを調べる検査を検討する必要がある。
着床前遺伝学的検査(PGT)
発端者の背景にあるメカニズムがUBE3Aの病的バリアントあるいはインプリンティングセンターの欠失にあることが判明している家系については、着床前遺伝学的検査が選択肢の1つとなりうる。(初期胚は相対的に低メチル化状態にあるため、これでDNAメチル化検査を行うことを前提としたPGTは問題が多い。)
その他
生殖補助医療(ART)
体外受精(IVF)ならびに顕微授精(ICSI)により、子にある種のインプリンティング障害(例えば、Beckwith-Wiedemann症候群)が生じる可能性が高まるというデータが存在する。2018年の報告では、IVF施行後のAS発生を調べることを目的として行った949の妊娠において、インプリンティングエラーに起因するASの発生が1例確認されている。この報告を行った研究者らは、IVFではインプリンティングエラーのリスクが高まる可能性があるとの仮説を立てているが、この研究は、試料数が少なく、研究に参加した出生前医療センターの数も多くないため、限定的なものとなっている[Johnsonら2018]。別の研究では、IVFやICSIとASとの間に有意の相関はみられないとのデータが出ている[Vermeiden & Bernardus 2013,Hattoriら2019]。
妊孕性
オランダとドイツの研究で、不妊の問題とASの発生との間に相関があることが示されている。AS罹患児を生むの段階で不妊の問題を抱えていたカップルの割合は、19%から25%に上っていた。ただ、イギリスでの調査では、不妊の問題とASとの間に正の相関は確認されなかった[Vermeiden & Bernardus 2013]。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- Angelman Syndrome Foundation, Inc. (ASF)
- Foundation for Angelman Syndrome Therapeutics (FAST)
- Medical Home Portal
- National Library of Medicine Genetics Home Reference
- NCBI Genes and Disease
- American Epilepsy Society
- Epilepsy Foundation
4255 Westbrook Drive
Suite 219
Aurora IL 60504
Phone: 800-432-6435 (toll-free); 630-978-4245
Fax: 630-978-7408
Email: info@angelman.org
www.angelman.org
PO Box 608
Downers Grove IL 60515
Phone: 630-852-FAST; 866-783-0078
Fax: 630-852-3270
Email: info@CureAngelman.org
www.cureangelman.org
Phone: 800-332-1000; 301-459-3700
Email: ContactUs@efa.org
www.epilepsy.com
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:Angelman症候群:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specificデータベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| UBE3A | 15q11.2 | ユビキチン-タンパク質リガーゼE3A | UBE3A database | UBE3A | UBE3A |
データは、以下の標準資料から作成したものである。遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:Angelman症候群関連のOMIMエントリー(内容の閲覧はOMIMへ)
| 105830 | ANGELMAN SYNDROME; AS |
| 601623 | UBIQUITIN-PROTEIN LIGASE E3A; UBE3A |
分子レベルの病原
Angelman症候群の中心的な症候は、母性継承のUBE3Aアレルの発現低下あるいは機能低下に起因して生じる[Jiangら1999,Lossieら2001,Nicholls & Knepper 2001]。UBE3Aは、ユビキチン-タンパク質リガーゼE3Aをコードする。このタンパク質はユビキチン化経路で作用し、標的タンパク質を分解する働きをする。UBE3Aは、ヒトの胎児の脳、ならびに成人の前頭野皮質内で母性優先の発現を示す[Rougeulleら1997,Vu & Hoffman 1997,Herzingら2001]。
UBE3Aは大きな5’CpGアイランドを有しており、母性アレルと父性アレルそのもののDNAメチル化の程度に差はみられない。UBE3A内にメチル化可変領域はなく、インプリンティングを受けたUBE3Aの発現は、父性発現のアンチセンス転写産物を通じて間接的な調節がなされている。Runteら[2001]は、AS/PWS領域には、SNURF-SNRPNのインプリンティングセンターに始まり少なくともUBE3Aの5’末端まで伸びる460kbの長いSNURF-SNRPNセンス/UBE3AアンチセンスRNA転写産物が存在することを明らかにした。このアンチセンス転写産物により、父性UBE3Aの発現がブロックされる仕組みになっている[Mengら2013]。
UBE3Aが破壊されると、本来であれば、ユビキチン-プロテアソーム系の働きにより平衡と機能が維持されるはずの、タンパク質の分解、置換、調節といった決定的に重要な神経細胞のプロセスに影響が及ぶ可能性がある。ユビキチン-プロテアソーム系は、シグナル伝達、細胞周期の進行、DNA修復、転写制御などの細胞機能に不可欠なものである[Ciechanover 1998,Hershko & Ciechanover 1998]。
病的バリアント
- 15q11.2-q13の欠失(65%-75%)
ASの発症に係わる15q11.1-q13の欠失イベントの大多数に、低コピー反復領域を特徴とする3つの切断点(近位のBP1,BP2と遠位のBP3)が関与している。これらの欠失のサイズは、おおむね5-7Mbである[Amos-Landgrafら1999,Christianら1999](図2参照)。15q11.2-q13欠失例の10%未満は、BP1/BP2から、より遠位にある低コピー反復領域であるBP4あるいはBP5までの欠失である(図2参照)[Sahooら2007]。
注:通常の欠失領域を外れた微小欠失として、BP1・BP2間の欠失[Doornbosら2009]、BP3・BP4間の欠失[Rosenfeldら2011]、ならびに、さらに遠位の15q13.3領域の関与する微小欠失症候群[Masurel-Pauletら2010]などの報告がみられる。しかしながら、これらの欠失の罹患者はASの症候を示さない。
- 15q11.1-q13のゲノム異常
健康に影響は表れてはいないものの、内在的に有しているゲノム異常が生殖細胞系列における15q11.1-q13の欠失の素因となり、結果としてASの子が生まれるといったことが生じうる。
- 15q11.1-q13の欠失に起因して生じたAS罹患児をもつ母親の一定割合が15q11.2-q13領域の逆位を有していることが判明している[Gimelliら2003]。
- 以前に、同一家系内の2人が欠失を有していた例(一方はPWSを、他方はASを引き起こす欠失)について、15q11.2-q13の染色体内逆位挿入の継承に起因したものとして報告された例がある[Collinsonら2004]。
- 15番染色体の父性片親性ダイソミー(3%-7%)
PWSとは対照的に、ASでみられる父性UPDは接合後起源であることが多い[Fridman & Koiffmann 2000,Robinsonら2000]。
減数分裂起源の父性UPDも確かにみられはするものの、このメカニズムは、PWSの際の母性UPDの場合ほど多くはない。
- インプリンティング異常(3%)
AS罹患者の中のこのサブセットは、配偶子形成時における正常なインプリンティングのリセットを障害するようなインプリンティング異常を有している。このタイプの罹患者は二親性継承の15番染色体を有してはいるものの、母親由来の15q11.2-q13領域は父性のエピゲノム型であるため、この領域にある母性発現遺伝子の転写は不活性となる[Buitingら2016]。
これらの例のインプリンティングセンター欠失のマッピング(ならびにPWS関連のインプリンティングセンター欠失のマッピング)により、インプリンティングセンター内の2つの重要エレメントであるAS-SRO、PWS-SROという2つの小欠失重なり領域(SRO;small regions of deletion overlap)の範囲が明らかとなった[Buitingら1995](図2参照)。PWS-SROは4.3kbのサイズで、SNURF-SNRPNのエクソン1/プロモーター領域と一部が重なる[Ohtaら1999]。AS罹患者のインプリンティングセンター欠失は、セントロメア寄りのSNURF-SNRPNのプロモーター/エクソン1領域に影響を及ぼす。
AS-SROは880bpのサイズで、SNURF-SNRPNのエクソン1より35kb近位に位置する[Buitingら2016]。
インプリンティング異常に起因してASに至った例の大多数は、ASインプリンティングセンターの欠失ではなく、インプリンティングセンターの機能を破壊するエピゲノムの異常を有している。
- UBE3Aの病的バリアント(11%近く)
フレームシフトを引き起こす小欠失や小重複、ミスセンス・ナンセンス病的バリアント、スプライシング異常、大欠失、複雑な再構成など、これまでに250を超える病的バリアントが報告されている。
更新履歴:
- 日本語訳者: 飯田崇博(信州大学医学部医学科),和田敬仁(信州大学医学部社会予防医学講座遺伝医学分野)
Gene Review 最終更新日: 2005.11.8. 日本語訳最終更新日: 2006.2.20. - Gene Review著者: Charles A Williams, MD, Aditi I Dagli, MD, Daniel J Driscoll, PhD, MD
日本語訳者: 窪田美穂(ボランティア翻訳者),鳴海洋子(信州大学医学部附属病院遺伝子診療部)
Gene Review 最終更新日: 2008.9.8. 日本語訳最終更新日: 2009.5.5. - Gene Review著者: Aditi I Dagli, MD,Charles A Williams, MD
日本語訳者: 窪田美穂(ボランティア翻訳者),石川亜貴(札幌医科大学医学部遺伝医学)
Gene Review 最終更新日: 2011.6.16.日本語訳最終更新日: 2014.3.7. -
Gene Reviews著者: Aditi I Dagli, MD, Jennifer Mathews, MS, CGC, and Charles A Williams, MD.
日本語訳者: 佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学) - GeneReviews最終更新日: 2021.4.22. 日本語訳最終更新日: 2023.5.28.
Gene Reviews著者: Aditi I Dagli, MD,Jennifer Mathews, MS, CGC,and Charles A Williams, MD.
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2021.4.22. 日本語訳最終更新日: 2023.7.30. [in present]
![]()