
AT/RT(atypical teratoid/rhabdoid tumor) ラブドイド腫瘍
非定型奇形腫様/ラブドイド腫瘍
embryonal tumor WHO grade 4
- AT/RTは日本語の適切な訳がありませんから,そのままAT/RT(エーティー・アールティー)と呼ばれます
- 胎児期の細胞から発生すると考えられています
- 乳児や幼児にできる腫瘍で3歳未満に多いです,赤ちゃんの脳腫瘍とも言えます
- 発症年齢中央値は1歳半くらいで,80-90%は3歳未満です
- 1歳未満の悪性脳腫瘍の50%ほどになります
- SMARCB1遺伝子産物 INI-1 (イニワン) に異常があります
- 小脳に多いのですが,脳のどの部分にも発生する腫瘍です
- 症状や画像などが似ているために髄芽腫やPNETと誤診されることがあります
- 頭蓋内圧亢進症状という頭痛や嘔吐,意識障害(反応がにぶい)で発症することが多いです
- 発症した時にはすでに巨大な腫瘍になっていることが多いです
- 髄液にのって脳の他の部分や脊髄に転移(髄液播種)します
- 診断はMRIでしますが,最初から必ず脊髄の方も調べなければなりません
- 3から4割で診断時にすでに播種が生じています
- 残念ながら悪性度が高くて治療も難しいものです
- 半数以上の子供たちが1年以内で亡くなります
- 2021年の時点で,世界的コセンサスがある治療方法(標準治療)はありません
- まず手術でできるだけとりますが,とり切れた時に助かる可能性が高くなります
- 手術で取り切れたと思ってもあっという間に再発(再燃)することがありますから,化学療法や放射線治療を急ぎます
- 化学療法といっても大量の制がん剤と髄注(髄液腔内へ薬を入れる)を使うものです
- 化学療法としては、IRS-IIIとかACNS0333のレジメンという方法に準じたものが期待されます
- 放射線治療ができた例で長期生存が報告されていますが,低年齢なので放射線障害も大きくでます
- 3歳未満の子供たちにはできれば放射線治療をしないで化学療法だけで治療するという選択肢がとられました
- でもそれでは助からないことも多いので,診断時に播種がない例では3歳未満の低年齢も含めて局所照射54グレイを選びます
- 局所照射といえども、年齢と腫瘍の部位によっては使えないこともあります
- 乳幼児を含めて長期生存例ではup-front radiation therapy(放射線治療からはじめる)がなされていたという報告があります
- 乳幼児の脳室内にありINI1蛋白異常があるものでは,良性の経過をたどる cribriform neuroepithelial tumor CRINET を考える必要があります
- 一方で,放射線治療を行うと知能予後(認知障害)がかなりつらい状態となるので,有効と解っていても使用しないという意見があります
- このように,治療法の選択には矛盾がいっぱいです
- あまりにも状態が厳しい場合では,何も治療しないで緩和ケアのみをするということも選択肢である脳腫瘍です
AT/RTのMRI画像

AT/RTのMRI像はさまざまです。乳幼児のgrade IVの悪性腫瘍ですから,増大速度が極めて速く,巨大で,腫瘍周辺脳浮腫が強く,ガドリニウムで強く増強されるというのが一般的です。硬膜を侵し頭蓋骨破壊を伴って頭蓋から外に膨隆してくるのもあります。多くは腫瘍内部に壊死が見られ壊死性のう胞が形成されます。CT/MRIで、腫瘍血流が豊富で細胞密度が高いという所見は重要な点です。
でもこの例(上の画像)の様に,mass effectも軽度で腫瘍周辺浮腫も目立たないものも珍しくはありませんし,他の胎児性脳腫瘍とは画像診断で鑑別することはできません。
治療の方向性
- 悪性度のとても高い腫瘍なので,助かるためには何でもしなければならないという考えにも立ちます
- 治療は一概には語れません,発生した年齢,脳の部位,播種転移があるかどうかで大きく変わってしまうからです
- 外科的立場からは,初期診断後に可能であれば開頭手術で全摘出します
- 放射線化学療法後に残存腫瘍があればそれを全摘出することを積極的に考慮します (second look surgery)
- 播種がある場合には,治療をしないで緩和ケアをだけをするというもの有力な選択肢です
AT/RTの化学療法
- かつての,Pediatric Oncology Group (POG)9233/4 と Children’s Cancer Group (CCG) 9921での2年EFSは6.4%と低いものでした
- 2020年時点で,Boston Protocol, EU-RHAB, ACNS0333などのプロトコールが用いられています
- MTXの髄注をしない,大量化学療法を用いる,脳脊髄照射ではなく可能であれば局所照射を用いるという方向性にあります
- 年齢,腫瘍の広がり,手術後の腫瘍サイズなどによって修飾 modificationされます
ACNS0333 protocolの概要
Reddy AT: Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report From the Children’s Oncology Group Trial ACNS0333. J Clin Oncol 2020
- 脳脊髄照射をしないで局所照射 local radiationを応用しようとする試みです
- 手術後に2コースの化学療法 (VCR, MTX, VP-16, CY, CDDP)
- MTX髄注は用いません
- その後に3コースの大量化学療法 (CBDCA, TT) と末梢血幹細胞救援
- 腫瘍床に原体照射を加えますが,病変の広がりと年齢によって時期と照射領域を変えます
- 3歳未満は50.4Gy,それ以上は54Gyです,陽子線の応用は許可されています
- 登録65人のうちの54人は3歳未満でした
- 2020時点では最も良い治療成績をだしています
- 4年EFS 37%,4年OS 43%でした
- 4割程度の子供達が長期生存できると考えていいでしょう
- 再燃が生じるとすれば91%が2年以内に生じました(2年再発しなければほとんど再発はないということです)
IRS-III ATRT protocolの概要
- まず,手術摘出あるいは生検してINI 1遺伝子の検索をします
- 放射線治療前化学療法をします
- 残存腫瘍がある場合には再手術を検討します
- M0(転移がない)あるいは3歳以下では原体局所照射 conformal irradiation と化学療法をしますが,M+(転移がある)あるいは3歳以上では脳脊髄照射と化学療法を行います
- 放射線治療後導入化学療法をします
- さらに残存腫瘍がある例では放射線外科 SRSが行えるかどうかを検討します
- 維持化学療法をします
- 脳脊髄照射がなされなかった例でのみドキソルビシン継続化学療法をします
WHO 2021年 の病理診断基準
- 組織像とINI1蛋白の欠失をみて診断します
- INI1蛋白発現は免疫組織染色で評価することができます
- chromosome 22の完全あるいは部分的欠失があります
- 結果として, SMARCB1gene (hSNF5/INI1)の異常(不活化)を生じます
- きわめてまれに,SMARKA4 (BRG1)の変異を有するものがあります
- もし組織診断でAT/RTであってINI1変異がない場合は,CNS embryonal tumor with rhabdoid featuresと記載します
- 免疫染色でINI1蛋白変異があり,SMARCB1欠失があっても,ラブドイド様の組織像見えない場合は,cribriform neuroepithelial tumor CRINET か desmoplastic myxoid tumor of the pineal region, SMARKB1-mutant を考えます
AT/RTのその他の知識
病理所見:European Rhabdoid Registryの記載より
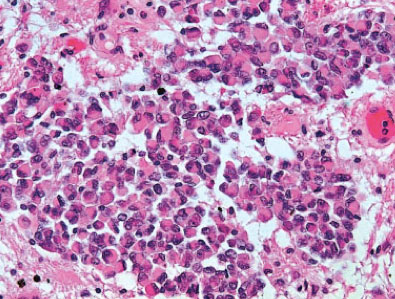
Macroscopically, AT/RT resembles medulloblastoma and sPNET. The tumors are soft, pale pink and show areas of hemorrhage as well as necrotic regions. Very commonly rhabdoid cells characterized by eosinophilic cytoplasm, large nuclei with excentric nucleoli and a prominent membrane as well as cytoplasmic eosinophilic inclusion bodies are seen. These diagnostic cells may be grouped in nests close to areas composed of neuroectodermal, mesenchymal or epithelial tissue types. Only about 10 to 15% of AT/RT consist almost exclusively of rhabdoid cells. AT/RT exhibit a broad spectrum of immunohistochemical reactions corresponding to the different tissue subtypes. Rhabdoid cells are characterized by expression of vimentin, EMA (epithelial membrane antigen) and cytokeratins, less commonly by SMA (smooth muscle actin). The immunohistochemical demonstration of lost INI1 protein expression in the tumor cells is a strong indicator for AT/RT, however, rare AT/RT with preserved INI1 expression are also on record.
細分類
- 分子遺伝学的にはさらに3−4つに分類されます
- ATRT-TYRは,最も幼い子(中央値12ヶ月)に発生し,テント下に多く,ガドリニウムで強く増強される wavy bandlike enhancementという特徴があります
- ATRT-SHH1はテント上,ATRT-SHH2はテント下に発生する傾向があり,両者ともにMRI所見はガドリニウム増強されない特徴があります
- ATRT-MYCは,年齢層が高く,テント上に発生し,周囲脳浮腫が極めて高度です
家族性AT/RT
Rhabdoid tumor predisposition syndrome (RTPS)
- 家族性のものは rhabdoid tumor predisposition syndrome (RTPS)ともいえわれます
- SMARCA4かSMARCB1 ( INI1, hSNF5) 遺伝子のgerm lineに異常がある家系が知られています
- 常染色体優性遺伝ですが浸透率は低いです
- myoepitheliomaやschwannomatosisを合併することがあります
- このタイプのAT/RTは,1歳未満の乳児の小脳に好発します
- 腎臓をはじめからだのどこにでも悪性ラブドイド腫瘍を合併あるいは同時発生する可能性があります
- でもしかし,de novo germline SMARCB1 mutationですから,実際にはほとんど遺伝しないのです
Bruggers CS, et al.: Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system.Pediatr Blood Cancer. 2011
ユタ大学ではAT/RT 20例のうち12例が家族性との診断でした。でもこの頻度は高すぎるでしょう。家族性のAT/RT 発症中央値生後5ヶ月,生存期間中央値5ヶ月と孤発性のものより予後が悪かったとのことです。
Bartelheim K, et al.: Improved 6‐year overall survival inAT/RT– results of the registry study Rhabdoid 2007.Cancer Med. 2016
ヨーロッパからのデータです。germ line mutationが21例中6例(29%)にあったとのことです。
AT/RTの学術情報
ACNS0333の結果 : 大量化学療法と三次元原体照射による治療
Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report From the Children’s Oncology Group Trial ACNS0333. J Clin Oncol 2020
3歳未満の54例の治療結果です。手術摘出後に、2コースの通常化学療法、3コースの大量化学療法、局所放射線治療で治療がなされました。放射線治療のタイミングは年齢と腫瘍の部位によって変えることができます。4年無増悪生存割合は37%、全生存割合は43%でした。放射線治療のタイミングは生存割合に影響しませんでした。再発は治療後2年以内に生じるとのことです。
SHHをさらに2型に分類できる
Ho B: Molecular subgrouping of atypicalteratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol. 2020
ATRT-SHH (sonic hedgehog), ATRT-TYR, ATRT-MYCに分類されてから5年,予後などが再検証されました。ATRT-SHHは,ATRT-SHH-1, ATRT-SHH-2にさらに細分化されるそうです。
epigeneticに3つに分類できる
Johann PD, et al.: Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell. 2016
Torchia J, et al.: Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell. 2016
Torchia J, et al.: Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol. 2015
Torciaの論文では259例,Johannの論文では192例のAT/RTが解析されました。3つのサブタイプに分類することができるとのことです。薬剤感受性や生存期間の長短も異なるとことですが,まだ臨床応用できるほどの知見とはなっていません。
ATRT-SHH (sonic hedgehog), ATRT-TYR (tyrosine), ATRT-MYC (myelocytomatosis oncogene)と呼称され,TYRはテント下に多い,MYCでは腫瘍周辺浮腫が強い,SHHではガドリニウム増強所見が29%ほどでみられない,などの画像上の特徴もあるようです。
cribriform neuroepithelial tumor CRINET
Johann PD: Cribriform neuroepithelial tumor: molecular characterization of a SMARCB1-deficient non-rhabdoid tumor with favorable long-term outcome. Brain Pathol 2017
20例の解析で,年齢中央値20ヶ月でした。全生存期間中央値は125ヶ月と10年を超えていました。
European Rhabdoid Registryからの報告:大量化学療法
Benesch M, et al.: High-dose chemotherapy (HDCT) with auto-SCT in children with atypical teratoid/rhabdoid tumors (AT/RT): a report from the European Rhabdoid Registry (EU-RHAB). Bone Marrow Transplantation 49, 370–375, 2014
年齢中央値生後21ヶ月の19人の子どもが登録されました。診断時に9人で播種転移の所見がありました。 手術全摘出は5人で可能でした。6コース程度の化学療法の後に大量化学療法 HDCT がなされ,14人では放射線治療が加えられました。HDCTの前に6人でCR,8人でPR,2人でSD,2人でPDとなっていました。追跡機間16ヶ月で14人に腫瘍進行 progressionが生じ,2年無増悪生存割合は29%,全生存割合は50%と算出されました。最終追跡時点では8人が生存していて,11人 (58%) が死亡しています。AT/RTの一部の患児では大量化学療法と放射線治療の効果があるかもしれないという結論です。結論を詳しく読むと,HDCTの後で3分の1の患児で継続的な寛解が得られるのですが,転移のない例 localized diseaseと導入化学療法が奏功した例と手術を含めて残存腫瘍が無いもしくは小さい例で有効であると記述されています。
「解説」使用された化学療法はRhabdoid 2007とEU-RHABです。Rhabdoid 2007は,VCD (VCR, CY, ADM), ICE (IFO, CBDCA, VP-16), itMTXのコンビネーションです。EU-RHABは,ADM, ICE, VCA (VCR, CY, actinomycin D), itMTXです。HDCTでは,CBDCA, thiotepaが用いられています。1コース目のICEの後でstem cell harvestが行われていますから,化学療法を開始する前にこの治療計画をしないとなりません。筆者らが結論しているように,導入化学療法が奏功しないとこの大量化学療法 HDCTに踏み込むべきではないとの解釈が妥当でしょう。
Head Start IIIの治療法は推奨できない
Zaky W, et al.: Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid /rhabdoid tumors: the Head Start III experience. Pediatr Blood Cancer 61: 95-101, 2014
Head Start IIIという治療計画に基づいて,19人のAT/RTの子どもが大量化学療法を基本として治療されました。放射線治療は原則として使用せず初期治療を開始しています。大量化学療法の回復を待って,年齢を考慮して,診断時の病変の広さを鑑みて,導入療法への反応をみてから使用されました。4人の子どもで導入化学療法を終えることができて,その内の3人でconsolidationができました。5人の子どもが治療の副作用で死亡しました (5 toxic deaths)。3年時点での全生存割合は26%でした。このまとめの時点で,3年以上生存していた子どもが4人 (21%) いたとのことです。
「解説」結果は燦々たる成績であり,これと同じ考え方の治療法はAT/RTに用いられるべきではないと言えます。
小児脳腫瘍先進国カナダからの情報
Lafay-Cousin L, et al.: Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 48: 353-359. 2012
カナダで1995年から2007年の12年間に,50人の子どもがAT/RTと診断されました。診断年齢中央値は生後16.7ヶ月で,52%はテント下発生でした。19人(38%)で転移がありました。10人は緩和ケアを受けました。治療を受けた40人の内,15人(30%)が全摘出を受け,22人が通常化学療法,18人が大量化学療法を受けました。髄腔内化学療法は9人です。15人が放射線治療を受けています。30人で腫瘍の進行悪化があり期間中央値は5.5ヶ月でした。生存期間中央値は13.5ヶ月,最長生存は117.5ヶ月で,年齢,腫瘍部位,播種は生存期間に関係ありませんでした。全摘出できた例の2年生存割合が60% (p=0.03) であったとのことです。大量化学療法 CBDCA/TTP での生存割合が良い傾向があり,放射線治療 upfront RTは影響がなく,12人の生存者のうち6人は放射線治療を受けていなかったとのことでした。
「解説」無増悪生存期間が半年に満たないということが確認されています。全摘出できたものは腫瘍の発生部位と大きさが外科適応となったことを示していますので,播種や部位が関係ないわけではありません。放射線治療を受けた15人の内の6人が生存しているとも解釈できます。
フランスからの情報
Dufour C, et al.: Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 118: 3812-3821, 2012
1998年から2008年までのフランスでの経験のまとめです。58人の小児が治療されました。診断の年齢中央値は1.4歳で,生後14日から8.5歳まででした。テント上が26例,テント下(小脳脳幹)が28例でした。INI1 nuclear expressionの欠損が50例中の49例(98%)に認められ,claudin-6が42例中37例(88%)で陽性となり,β-catenin, P53なども半数以上で陽性でした。初期治療は47人が化学療法で,16人が放射線治療でした。生存期間中央値は9ヶ月とかなり厳しい結果でした。2歳以下,播種転移があった例,claudin-6の染色性が強い例で有意に予後が悪かったそうです。
大量化学療法
Finkelstein-Shechter T, et al.: Atypical Teratoid or Rhabdoid Tumors: Improved Outcome With High-dose Chemotherapy. J Pediatr Hematol Oncol 32: 182–186, 2010
トロント子ども病院からの報告です。2003-2008年に8人の子どもがAT/RTと診断されました。5人ですでに髄液播種があったそうです。生後1ヶ月と6ヶ月の2人の乳児は治療を受けず,6人の子どもに手術治療と大量化学療法(幹細胞移植)が行われました。局所放射線治療を受けた子どもは2人です。経過観察期間中央値52ヶ月の段階で4人が無増悪生存していましたが,全員で認知機能障害がありました。播種がある状態で発症した2人では化学療法のみで長期生存が得られています。
手術後の基本的な化学療法は3 cycles of VP-16, cyclophosphamide, cisplatin and vincristineで,大量化学療法はcarboplatin-thiotepa conditioning regimenでした。
AT/RTの情報
Tekautz TM, et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23: 1491-1499, 2009
2009年のJ Clin Oncolという権威のある雑誌に載った論文です。 31例中22例が3歳未満であり,全員が手術摘出を受けて30人が化学療法を受けました。2年全生存割合は3歳未満で17%,3歳以上では89%と大きな差があったとのことです。3歳以上の患児では術後放射線治療と大量のアルキル化剤の投与で治る可能性があると書かれています。また小数例ですがICE化学療法 (ifosfamide, carboplatin, etoposide) が有効であったとのことです。一方,多くの患児が3歳未満で発症しますが,その年齢層で2年の時点での死亡割合は8割を超えると理解しなければなりません。また3歳未満の例では化学療法の最中に腫瘍再燃(増大進行)してしまうし,診断からの無増悪期間中央値は4ヶ月にしかならず,現時点では有効な化学療法のレジメンは見あたらないと考察されています。長期生存例では乳幼児も含めてup-front radiation therapyがされているとのことです。
AT/RTの情報
Chi SN, et al.: Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27: 385-389, 2009
下に書いてあるChiという先生の学会発表の結果が論文になったものです。2004年から2006年まで25例のAT/RTの子供が治療されて20人結果がまとめられています。年齢中央値は2歳2ヶ月,14例 (70%) では播種がなく,11例で全摘手術がされました。15人が放射線治療を受けていますが,年齢と播種があるかないかで放射線治療が選択され,局所照射54グレイ(11人)のみか脳脊髄照射36グレイ(4人)が加えられています。2年無増悪生存割合は53%で,全生存割合は70%でした。照射前化学療法はmodified IRS-III regimenが使用され,その後にchemoradiation induction, postradiation induction, maintenance, continuation, intrathecal therapyなどの複雑な治療法が組まれていますので原著を読まなければ解りません。 評価可能であった12例の照射前化学療法の有効性 (objective response) は58%でした。この論文を詳しく読むと半数近くの子供で治る可能性がある治療ができたということになります。
AT/RTの情報
Gardner SL, et al.: Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatr Blood Cancer 51:235-240, 2008
ニューヨーク大学からの報告です。13人の子どもが,Head Start Iか Head Start IIという化学療法の組み合わせで治療されました。5コースのcisplatin, vincristine, cyclophosphamide, etoposide通常化学療法。さらに1997年からのHS IIでは,メソトレキセート大量化学療法が加えられました。地固め大量化学療法はcarboplatin, thiotepa, etoposideで,自家血液幹細胞救援がなされました(AHPCR)。HS IIで治療を受けた7人中3人 (43%) の子どもが放射線治療を受けずに生存していたそうです(治療後42ヶ月,54ヶ月,67ヶ月です)。この治療方法を真似ようにも,残念ながら日本ではtiotepaが使えません。
AT/RTの学会発表
Results from a single-arm multiinstitutional study of multiagent intrathecal and systemic chemotherapy with age- and risk-adjusted radiation therapy for childern with newly diagnosed CNS atypical teratoid./rhabdoid tumor (DFCI 02-294). Chi S, et al. ISPNO 2008, Chicago
2004年から2007年にmodified IRS-III regimenという方法で治療された22人の患児の報告です。とても大まかには,6週間の導入化学療法の後で,放射線治療(化学療法併用)がされて,さらに地固め科学療法が6週間,維持化学療法が 26週間,追加の化学療法をさらに6週間する方法です。制がん剤の髄腔内投与も行われました。転移のない例(M0)では局所照射confomal RTを54グレイしてます。3歳以上で転移(播種)のある例では36グレイの脳脊髄照射がされています。年令の中央値は2.5歳,テント上が12例で,テント下が10例でした。半分の患児でおよその全摘出がなされています。16例(73%)で転移がないM0というstageでした。予定の治療がすべて完遂できたのは22人中の12人であり,放射線治療を受けたのは17人でした。放射線前の化学療法奏功率(CR+PR)は62%という高い数字です。2年の時点での無増悪生存割合は48%で,全生存割合は67%でした。化学療法の副作用は当然強いもので,化学療法死が1例あったとのことです。
この報告をみる限り,ほとんど救うことのできないとされたAT/RTの子供たちにも生存の機会はあると思えます。
AT/RTの学会発表
CNS ATYPICAL TERATOID RHABDOID TUMOR (ATRT) IN CHILDREN LESS THAN 36 MONTHS: A CANADIAN PEDIATRIC BRAIN TUMOR CONSORTIUM (CPBTC) EXPERIENCE. Lafay-Cousin L, et al. ISPNO 2008, Chicago
カナダでの調査です。3歳以下の脳腫瘍の子供531人のうちの24人(4.5%)がAT/RTでした。平均診断時年令は12.6ヶ月です。テント上が33%,テント下が58%, 脊髄が8%です。24人中の10人(42%)で診断時からすでに播種転移がありました。8人が手術だけ,7人が化学療法のみを受け,9人が放射線化学療法を受けています。腫瘍が治まっていた無増悪生存期間中央値は9ヶ月,外科手術のあとで追加治療を受けた16人の生存期間中央値は16ヶ月でした。1年生存割合50%,2年生存割合は19%です。90%以上の切除を受けた子供の生存期間がながかったようですが,放射線治療と大量化学療法は予後に寄与しなかったとしています。結果的に3人の子供が生存していて,その生存期間は,20, 21, 78ヶ月だそうで,これらの子供は90%以上の切除がされて,1例は放射線治療を受けて,2例は大量化学療法を受けていました。
AT/RTの学会発表
German pediatric atypical teratoid/thabdoid tumors: Interim analysis of the ATRT-CNS study and retrospecitve analysis. Peters O, et al. ISPNO 2008, Chicago
ドイツからの19症例の治療報告です。1例で完全摘出,5例で亜全摘,9例で部分摘出,4例で生検術がなされました。手術後に,2コースの導入化学療法(doxorubicin, dactinomycin, cisplatin, vincristine, methotrexate)を行った後に,54Gyの局所照射とcarboplatinの併用がされ,さらに最初の化学療法とおなじものが1コース投与されました。 地固め療法として6コースのCCNU, cisplatin, vincristine, methotrexate)が追加されています。無増悪および全生存割合は79%であり,生存期間の平均値は50ヶ月と報告されました。 最初の2コースの化学療法の奏功率は,1 continued complete response, 2 complete response, 13 partial response, 3 stable diseaseとかなり高いものです。有害事象の際立ったものは粘膜障害と感染症であったと記録されていますが,目立った脳脊髄障害は報告がなかったということです。結論として,AT/RTは化学療法に感受性のある腫瘍であるが,化学療法の有害事象はかなり強いとされています。
AT/RTの総論
Squire SE, et al. Atypical teratoid/rhabdoid tumor: the controversy behind radiaiton therapy. J Neurooncol 81: 97-111, 2007
- AT/RTは小児脳腫瘍の2-3%
- 髄芽腫に応用される治療では治らない
- 手術摘出と化学療法のみではかなりの例で局所再燃が生じている
- 長期生存例は,初期治療に大量化学療法と放射線治療を応用した時に期待できる
- vimentin, EMA, SMAの陽性率が高い
- 第22染色体22q11 の異常が高率にある
- complete resectionがoverall survivalに有意に影響する
- Intergroup Rhabdomyosarcoma III Study (IRS-III) のガイドラインで治療され長期生存した例が散見される
- high-dose platinum-based or alkylator-based chmotherapyが応用されることが多い
- 放射線治療の基本的役割は局所再発を防ぐことであるが,脳脊髄照射をとりいれるregimenが多い
- 診断後,3歳未満の低年齢児であろうともできれば早期の放射線治療が必要であろう
- 低年齢児のために局所放射線治療に止めざるを得ないという考え方もある
- 再発の1/3から1/2は髄液播種である
- 腫瘍床には50-55Gy程度,脳脊髄には23-30Gyを用いることが多い
- photon-IMRTの有用性はわからない
- radiosurgery (SRS) で局所を治療できれば有効であろう
最初の論文
1985年にBrinerによってはじめて記載されました。Briner, J., et al.: Malignant small cell tumor of the brain with intermediate filaments – a case of a primary cerebral rhabdoid tumor. Pediatric Pathology 3 : 117-118, 1985
当時はINI-1が知られていませんでしたので、光顕病理での形態診断でしたから,rhabdoid cellが目立たないとAT/RTと診断されません。1990年代ではこの診断基準によっての死亡割合は100%に近い,余命は6ヶ月以内と言われたこともありました。どんな治療をしても死亡する悪性脳腫瘍であると捉えられたのです。その後にINI1変異によっての診断が優先されるようになり,rhabdoid cellが優位でなくてもAT/RTと病理診断されるようになりました。かつての腫瘍群とはことなったcohortである可能性を含む,この診断基準の変化によって,長期生存例が20%を越えるという変化が生じました。もちろん画像診断,手術,化学療法の進歩も生存割合の向上に寄与しています。
文献
- Benesch M, et al.: High-dose chemotherapy (HDCT) with auto-SCT in children with atypical teratoid/rhabdoid tumors (AT/RT): a report from the European Rhabdoid Registry (EU-RHAB). Bone Marrow Transplantation 49, 370–375, 2014
- Briner, J., et al.: Malignant small cell tumor of the brain with intermediate filaments – a case of a primary cerebral rhabdoid tumor. Pediatric Pathology 3 : 117-118, 1985
- Chi SN, et al.: Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27: 385-389, 2009
- Dufour C, et al.: Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 118: 3812-3821
- Finkelstein-Shechter T, et al.: Atypical Teratoid or Rhabdoid Tumors: Improved Outcome With High-dose Chemotherapy. J Pediatr Hematol Oncol 32: 182–186, 2010
- Gardner SL, et al.: Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatr Blood Cancer 51:235-240, 2008
- Lafay-Cousin L, et al.: Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 48: 353-359. 2012
- Squire SE, et al. Atypical teratoid/rhabdoid tumor: the controversy behind radiaiton therapy. J Neurooncol 81: 97-111, 2007
- Tekautz TM, et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23: 1491-1499, 2009
- Zaky W, et al.: Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid /rhabdoid tumors: the Head Start III experience. Pediatr Blood Cancer 61: 95-101, 2014
