神経線維腫症1型
(Neurofibromatosis 1)
[Synonyms:NF1, Von Recklinghausen Disease, Von Recklinghausen’s Neurofibromatosis]
Gene Reviews著者: J M Friedman, MD, PhD
日本語訳者: 佐藤康守(たい矯正歯科),森川真紀(名古屋大学医学部附属病院ゲノム医療センター),生田国大(名古屋大学医学部附属病院整形外科)
GeneReviews最終更新日: 2022.4.21. 日本語訳最終更新日: 2023.11.20.
要約
疾患の特徴
神経線維腫症1型(NF1)は多発性のカフェ・オ・レ斑,間擦部の雀卵斑様色素斑,多発性の皮膚神経線維腫,学習障害や行動障害などの所見が複数の器官系にみられる疾患である.NF1患者の約半数に叢状神経線維腫がみられるが,体内に生じ無症状であることが多い.叢状神経線維腫によって,疼痛,神経障害,病変部や隣接部の構造異常が生じることがある.頻度は低いもののより重症化しやすいものとして,視神経や中枢神経系の神経膠腫,悪性末梢神経鞘腫瘍,側彎症,脛骨異形成症,血管病変,消化器疾患,内分泌疾患,肺疾患がある.
診断・検査
発端者におけるNF1の診断は,臨床的特徴の2つ以上を有するか,もしくは1つの臨床的特徴に加えてNF1遺伝子のヘテロ接合性病的バリアントを有することで確定する.
臨床的マネジメント
症状に対する治療:
眼 や中枢神経系,末梢神経系,心血管系,肺,内分泌系,脊椎,長管骨の異常に対する治療に関しては,その分野を専門とする医師に紹介する.皮膚や皮下にみられる散在性の神経線維腫の中で,美容上問題となるものや不快な症状のあるものについては,外科的切除をおこなう.びまん性や大きな叢状神経線維腫については,外科的治療も可能ではあるものの,病変部の神経や隣接組織の損傷につながる可能性があるばかりでなく,取り残した腫瘍の成長を促しかねない.悪性末梢神経鞘腫瘍は可能であれば外科的に完全切除することが治療法として選択されるが, 症例によっては化学療法が有効なこともある.視神経膠腫は通常無症状で臨床的に安定しているため,特に治療を要しないことが多い. dystrophic typeの側彎症は外科的治療を要することが多いものの,non-dystrophic typeの側彎症は通常保存的治療で十分に対応可能である.発達や学習面の問題については個別に対応していくことが有益と思われるが,注意欠陥多動性障害の小児に対してはメチルフェニデートによる治療が有効とされる.
サーベイランス:
- 本症に精通した医師による身体診察を年に一回実施
- 眼科診察を小児では年に一回実施,成人では頻度は少なくてよいものの定期的に実施
- 小児の発達評価
- 定期的な血圧測定
- 頭蓋内腫瘍やその他の腫瘍が臨床的に疑われるものの診察だけではわかりづらい場合は,MRI検査による病変の同定とフォローアップ.
- 女性は30歳から年1回のマンモグラフィー検査を開始,更に30-50歳の女性では年1回の乳房MRI検査も検討.
- NF1遺伝子全体の欠失を有する患者,大きいあるいは増大する叢状神経線維腫や頭蓋内腫瘍を有する患者,症候性の血管障害や進行性の骨病変を有する患者、その他深刻な症候がみられる患者については, 症候に合わせたフォローアップをより頻繁におこなう必要がある.
遺伝カウンセリング
NF1は常染色体顕性遺伝(優性遺伝)の形式をとる.患者の約半数はNF1遺伝子の新生(de novo)の病的バリアントに起因する.NF1患者の子はそれぞれ50%の確率で病的バリアントを受け継ぐ.浸透率はほぼ100%である.したがって,NF1の病的バリアントを受け継いだ子にはNF1の所見が現れることになるものの,その重症度は罹患者である親よりも重度なこともあれば軽度なこともある.リスクのある家系員に対する出生前診断や着床前診断は,家系内で病的バリアントが同定されていれば可能である.
訳注:日本では, 本症に対する出生前診断や着床前診断は行われていない. いずれにしても次世代への遺伝に関しては細心の遺伝カウンセリングが必要である.
診断
本疾患を示唆する所見
下記の臨床症候のうちいずれかを有する患者は,神経線維腫症1型(NF1)を疑うべきである.
- 思春期前では最大径5mm以上,思春期以降では最大径15mm以上のカフェ・オ・レ斑(図1)が6個以上
- 腋窩や鼠径部の雀卵斑様色素斑
- 神経線維腫(図2)が種類を問わず2個以上,あるいは叢状神経線維腫(図3)が1個
- 視神経膠腫
- 細隙灯顕微鏡で同定された虹彩小結節(Lisch結節)が2個以上,あるいは脈絡膜の異常(光干渉断層画像化法や眼底の近赤外線反射イメージングでみられる明るく斑状の結節)が2個以上
- 蝶形骨異形成や脛骨前外側の弯曲,長管骨の偽関節形成などの特徴的骨病変
- 片親がNF1の診断基準を満たしている
- NF1遺伝子の生殖細胞系列病的バリアントを有している
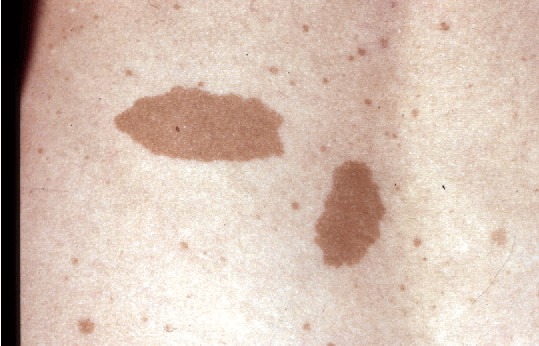
図1.カフェ・オ・レ斑
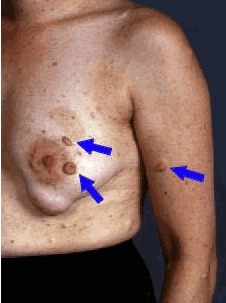
図2.神経線維腫
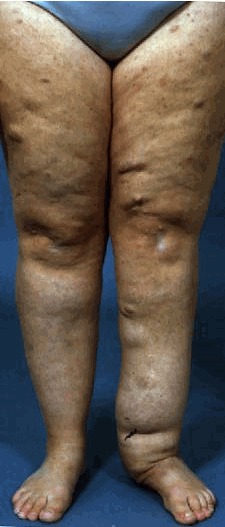
図3.叢状神経線維腫
発端者におけるNF1の診断は,前項の「本疾患を示唆する所見」に記された症候のうち2つ以上該当すれば確定する[Legius et al 2021].
注釈:
- Legius症候群患者(「鑑別診断」の項を参照)の約半数は,NF1の診断基準に合致する皮膚の色素所見(すなわち6個以上のカフェ・オ・レ斑,および腋窩部や鼠径部の雀卵斑様色素斑)を有する.しかし,Legius症候群患者の場合は,神経線維腫,視神経膠腫,虹彩小結節,NF1に特徴的な骨病変といったものがみられない[Legius et al 2021].さらに言うと,Legius症候群はNF1よりはるかに稀な疾患で,Legius症候群の小児患者の片親はほぼ例外なくカフェ・オ・レ斑と間擦部の雀卵斑様色素斑のみを有している(「鑑別診断」の項を参照).
- NF1においては,カフェ・オ・レ斑,間擦部の雀卵斑様色素斑,皮膚の神経線維腫,虹彩小結節が通常両側性に出現する.そうした病変が片側のみ,あるいは体の一部分のみに現れるといった場合は,モザイク型のNF1である可能性を考慮する必要がある.
- 蝶形骨翼の異形成と眼窩の叢状神経線維腫が同側にみられる場合は,診断基準にある個々の項目として勘定することはせず,両方で1つとみなす.
- NF1遺伝子の病的バリアントを診断基準の1つとできるのは,遺伝学的検査で生殖細胞系列の病的バリアントが確認された場合のみである[Legius et al. 2021]. 腫瘍組織のみでNF1遺伝子の病的(pathogenic)バリアントが同定された場合であるとか,生殖細胞系列であっても病的疑い(likely pathogenic),あるいは病的意義不明なバリアント(variant of uncertain significance)が同定された場合などは,診断基準の1つを満たしたことにはならない.
- 分子遺伝学的検査でNF1遺伝子の病的バリアントが検出されなかったとしても,NF1の診断が否定されるわけではない[Accettoro et al.2020].臨床診断基準に基づいてNF1と診断された患者の中には,現在の技術では病的バリアントが検出されない例も存在する.NF1の臨床的特徴の多くは年齢とともに出現頻度が増える傾向にあるため,成人期には疑いの余地なくNF1と診断できる患者の中にも,幼少期にはまだ所見が明らかには認められず,診断にまで至らないような例がみられる.
表現型からNF1の可能性が示唆される場合には,単一遺伝子検査を検討する.
NF1遺伝子のゲノムDNA(gDNA)やcDNA(mRNAから逆転写された相補的DNA)のシークエンス解析は,標的遺伝子欠失解析と共に実施される.スプライスに影響を及ぼす病的バリアントの頻度が高い(22-30%であり,そのうち三分の一以上はタンパク質コード領域に対するゲノムDNA解析では検出されない)ことから,cDNA解析を含む方法はゲノムDNA解析単体に比べて検出率が高い.(表1)
注釈:
- NF1の色素所見のみを有する患者でNF1遺伝子に病的バリアントが検出されなかった場合には,SPRED1遺伝子(「鑑別診断」の項を参照)のシークエンス解析ならびに欠失/重複解析が考慮される.
- 染色体マイクロアレイ解析(CMA)は,臨床所見からNF1遺伝子の微小欠失が疑われる場合にNF1遺伝子全欠失も検出できるようシークエンス解析の代わりに実施することがある.(「遺伝子型と表現型の相関」参照)
- 染色体の核型分析は,NF1の臨床診断が確実であるにもかかわらず,NF1遺伝子のゲノムDNAやcDNAのシークエンス解析,および標的遺伝子欠失解析からは病的バリアントが検出されなかった場合に,転座や複雑な細胞遺伝学的異常を調べる目的において考慮されることがある.
過剰な色素沈着や腫瘍,あるいはその他の特徴的所見が共通していて表現型からは他の疾患と区別できないような場合は,NF1やSPRED1,その他関連遺伝子(「鑑別診断」の項を参照)を含んだマルチジーンパネルが考慮されることがある.通常はRAS/MAPK症候群を対象としたマルチジーンパネルが最も適している.
注釈:
(1)パネルに含まれる遺伝子の種類や,遺伝子毎の検査の診断感度は,検査機関によって異なり,経時的に変化する可能性がある.
(2)マルチジーンパネルによっては,GeneReviewで述べる疾患と無関連な遺伝子が含まれることがある.そのため,本来の表現型からは説明できない遺伝子の病的バリアントや病的意義不明なバリアントが同定されることを考慮しつつ,臨床医はどのマルチジーンパネルが疾患の遺伝的要因を特定できる可能性が高いかを見極めなければならない.
(3)検査機関によっては,独自のカスタムメイドパネルや,臨床医が解析遺伝子を選定できる表現型中心のカスタムメイドのエクソーム解析といったオプションを提供している.
(4)パネル検査で用いられる手法には,シークエンス解析,欠失/重複解析,またはシークエンス以外の手法を用いた検査が含まれうる.
マルチジーンパネルの概要はここをクリック.遺伝学的検査を依頼する臨床医向けのより詳細な情報についてはこちらを参照のこと.
表現型が非特異的な場合(例えば,幼児期における発達遅滞や筋緊張低下)は,網羅的な遺伝学的検査(エクソーム解析やゲノム解析)を検討する.
注釈:NF1遺伝子の病的バリアントを有する例についても,特徴的な臨床症候(「本疾患を示唆する所見」の項を参照)を少なくとも1つ以上有していることが確定診断の条件となる.
網羅的な遺伝学的検査の概要はここをクリック.遺伝学的検査を依頼する臨床医向けのより詳細な情報についてはこちらを参照のこと.
表1 :神経線維腫症1型で用いられる分子遺伝学的検査
| 遺伝子1 | 検査法 | この方法により検出可能な病的バリアント2を有する割合 |
|---|---|---|
NF1 |
cDNAおよびゲノムDNAを用いたシークエンス解析3,4 | >95%5 |
| ゲノムDNA単独のシークエンス解析3 | ~60%-90%6 | |
| 標的遺伝子欠失/重複解析4 | ~13%7,8 | |
| 染色体マイクロアレイ解析(CMA)9 | ~5-11%7,8 | |
| 染色体核型分析 | <1%9 |
- 染色体座位とタンパク質については,表A.「遺伝子とデータベース」参照
- 当該遺伝子で検出されるバリアント情報については,「分子遺伝学」の項を参照
- シークエンス解析で検出されるバリアントは,良性, おそらく良性,病的意義不明,病的疑い,病的の5つに分類される.検出されるバリアントの種類としては,遺伝子内の小さな欠失/挿入,ミスセンス,ナンセンスおよびスプライス部位のバリアントが含まれる.NF1遺伝子の分子遺伝学的解析では,欠失/重複解析も含まれることが多い.エクソーム解析が用いられる場合には,欠失/重複解析(通常は次世代シーケンサーの読み取り深度のデータ解析による)が含まれているかを確認する. シークエンス解析結果の解釈について考慮すべき課題については,ここをクリック.
- 標的遺伝子の欠失/重複解析では, 遺伝子内の欠失や重複が検出される.シークエンス解析の際に欠失/重複解析(例:読み取り深度の解析)が実施されない場合は,代替法として定量PCRや,MLPA(multiplex ligation-dependent probe amplification)法,FISH法が考えられる.
- Evans et al [2010],Sabbagh et al [2013],Giugliano et al[2019]
- Maruoka et al [2014], van Minkelen et al [2014], Pasmant et al [2015], Zhang et al [2015], Calì et al [2017] ,Bianchessi et al[2020].
- 遺伝子の全欠失はNF1患者の5-11%にみられる.[Kehrer-Sawatzki et al 2020].
- Stenson et al[2020].
- オリゴヌクレオチドやSNPを用いた染色体マイクロアレイ検査(CMA)では,シークエンス解析では検出されないような(NF1遺伝子を含む)ゲノムワイドの大規模欠失/重複が検出される.同定可能な欠失/重複のサイズは,使用するマイクロアレイの種類や17q11.2領域のプローブの密度による.現在臨床で用いられている染色体マイクロアレイ検査は17q11.2領域も対象として設計されている.
臨床的特徴
臨床像
神経線維腫症1型(NF1)はきわめて多彩な多臓器疾患である.患者個人の生涯においても,同じNF1遺伝子病的バリアントを有する家系員の間においても,症状の進行や重症度は異なる[Gutmann et al 2017,Monroe et al 2017,Gianluca et al 2020].NF1の主要な臨床症候としては,多発性のカフェ・オ・レ斑,間擦部の雀卵斑様色素斑,多発性の皮膚神経線維腫,皮下ないし深部の結節性神経線維腫,叢状神経線維腫,特徴的な眼症候などがある.NF1患者においては,学習障害,行動障害,適応障害も非常に多くみられる.視神経膠腫や視神経以外の神経膠腫の頻度は比較的高いものの,そうした腫瘍の大多数は良性の経過をたどる.特に叢状神経線維腫や深部の結節性神経線維腫を多数あるいはサイズの大きいものを有するNF1患者は悪性末梢神経鞘腫瘍の発症リスクが高く,一般集団よりはるかに若く発症し,予後も不良であることが多い.NF1の女性は乳癌の発症リスクが高く,妊娠時の合併症も通常より多くみられる.NF1患者では高血圧が多くみられ,小児や若年成人患者においては,NF1の血管病変が脳卒中やその他の心血管合併症を惹起する可能性がある.患者によっては,脊椎や脛骨の異形成が大きな障害となることがある.また,頻度としては比較的低いものの,NF1関連の消化器系,内分泌系,あるいは肺の疾患がかなり深刻になることもある.
表2: 神経線維腫症1型における主な症候と出現頻度
| 症候 | 症候を有する患者の割合1 | 典型的な発症年齢 | 備考 |
|---|---|---|---|
| カフェ・オ・レ斑 | >99% | 乳児期・小児期 | 生後数年間で数が増加しサイズが増大する; 高齢になると消退する. |
| 間擦部の雀卵斑様色素斑 | 85% | 幼児期・小児期早期 | 小児期には年齢とともに頻度が上昇する |
| 虹彩小結節(Lish結節) | >95% | 小児期早期 | 小児期には年齢とともに頻度が上昇する |
| 脈絡膜の異常 | 82%-98% | 小児期早期 | 小児期に数が増加しサイズが増大する |
| 視路の神経膠腫 | 15%-20% | 出生時から6歳 | 成人では低頻度 |
| 視路以外の神経膠腫 | 2%-5% | 年齢と無関係 | 小児は成人より低頻度 |
| 皮膚の神経線維腫 | 99% | 思春期から成人期 | 小児期では稀; 生涯を通じ,大きさや数の変動が大きい. |
| 結節性神経線維腫 (皮下ないし深部) |
~15% | 思春期 | 左記の頻度は臨床検査に基づく; 全身MRI検査では2-3倍高くなる |
| 叢状神経線維腫 | ~30% | 乳児期(時に出生時)または小児期 | 左記の頻度は臨床検査に基づく; 全身MRI検査による頻度は~50% |
| 悪性末梢神経鞘腫瘍(MPNST) | 8%-13% | 思春期から成人期 | 小児期中期以降の横断的な有病率は2%-5%. |
| 知的障害 | 4%-8% | 小児期 | 生涯持続する |
| 学習障害 | 50%-60% | 小児期 | 生涯持続する |
| 行動障害 | 30%-67% | 小児期 | |
| てんかん | 6%-7% | 年齢と無関係 | |
| 長骨の異形成 | 2% | 乳児期(出生時) | |
| dystrophic typeの 側彎症 |
5% | 6-10歳 | 椎骨異形成により急速に進行する側彎 |
| non-dystrophic typeの側彎症 | 5% | 思春期 | 椎骨の異常を伴わない軽度の側彎. |
| 骨粗鬆症 | ~20% | 中年期 | 骨減少症は全年齢で多くみられる. 骨粗鬆症は一般集団より早期に発症するものの,小児や若年成人では稀 |
| 高血圧 | ≧15%-20% | 年齢と無関係 | 有病率は小児より成人のほうが高い. |
- この表に記載された症候の多くは,年齢ごとに出現頻度が異なる.この表は生涯の累積発症頻度を示しているため,ある一時期における有病率に比べて,はるかに高い数字になっていることがある.この表に記載された数値の大部分はFerner & Gutmann [2013]あるいはDeBellaら[2013]から引用したものである.近年判明した内容については,以下に記載する各症候の項で参考文献を示している
皮膚の特徴
カフェ・オ・レ斑(CALMs)
NF1患者にみられる特徴的なカフェ・オ・レ斑(CALMs)は,通常大きさが1~3cm程度の境界明瞭な長円形で,色合い(本来の皮膚の色よりやや濃い)は均一である.しかしながら,小さいものや大きいもの,淡い色合いや,より濃い色合いのもの,いびつな形のものもみられる[Ozarslan et al 2021,Albaghdadiet al 2022].不規則な色素沈着として,典型的な大きめの色素病変の中に雀卵斑様色素斑や小さく色の濃いCALMsがみられることもある.CALMsは通常乳児期や小児期早期からみられ,いったん形成されるとその数や大きさは変化しない(皮膚そのものの成長による拡大は除く).高齢のNF1患者については,加齢に伴う皺やびまん性の色素沈着のために見えにくくなることがある.
CALMsは扁平で周囲の皮膚と同じく平面的である.皮膚病変が盛り上がっている,あるいは周囲の皮膚に比べて柔らかく手触りが異なる場合は,皮下の叢状神経線維腫が考えられる.色白や浅黒い肌の人のCALMsは,色がやや濃いものの周囲の皮膚の色に似ていて見つけにくいことがある.そのような場合,色素斑を明らかにするのにウッド灯が有用である.CALMsはNF1患者の手掌や足底にはみられないものの,それ以外の全身に生じうる.
雀卵斑様色素斑.
多発する雀卵斑(そばかす)は日光に曝される部位によく生じるが,NF1患者では体幹や近位四肢,頸部にも散在的にみられることがある. NF1患者では腋窩や鼠径部,女性では乳房の下部といった皮膚同士が擦れ合う部位に雀卵斑様色素斑が生じやすい[Ozarslan et al 2021,Albaghdadiet al 2022].
神経線維腫. 「その他の腫瘍」の項を参照
その他の皮膚所見.
若年性黄色肉芽腫や貧血母斑はNF1患者に比較的よくみられるため,診断基準を満たさない幼児の診断の補助として役立つことがある[Miraglia et al 2020, Ozarslan et al 2021].若年性黄色肉芽腫は小さな黄褐色またはオレンジ色の丘疹で多発することがある.貧血母斑は不規則な形状の斑点で,周囲の皮膚よりも淡く,こすっても周囲の皮膚のように赤くはならない.
眼所見
虹彩小結節(Lisch結節)
虹彩小結節(Lisch結節)は無害な虹彩過誤腫で,細隙灯顕微鏡を実施するとほぼすべての成人NF1患者にみられるが,5歳未満のNF1患者では半分以下にしか認められない.虹彩小結節は三次元的な結節状の外観をもつことから,虹彩斑との判別が可能である.
脈絡膜色素斑
脈絡膜色素斑は一般的な眼科検査では検出されないが,走査型レーザー検眼鏡や近赤外線反射イメージング,あるいは光干渉断層画像化法(OCT)によるスキャンで可視化することができる[Vagge et al 2016, Moramarco et al 2018].軸索を同心円状に取り囲むシュワン細胞の増殖は,あらゆる年代のNF1患者の多くに認められるが,小児期においては有病率の上昇,数の増加,サイズの増大がみられる[Touzé et al 2021].
他の眼所見
NF1患者の眼科検査でしばしば確認されるその他の所見としては,網膜内細小血管異常[Parrozzani et al 2018,Moramarco et al 2019]や眼底の斑状の色素沈着[Moramarco et al 2021]などがある.
視路神経膠腫
NF1患者にみられる視路神経膠腫は通常無症状で,生涯を通じて無症状のままである[Di Nicola & Viola 2020,Shofty et al 2020].視路神経膠腫の臨床経過は,NF1でない患者に比べてNF1患者の方が軽度な傾向がある.NF1による視路神経膠腫は長年にわたって変化しないか,もしくは非常にゆっくりと進行することが多い[Sellmer et al 2018,Kinori et al 2021].さらに,視路神経膠腫の多くは自然に消退していくため,有病率は幼児期では約20%だが高齢のNF1患者では5%未満にまで低下する[Sellmer et al 2018].症候性の視路神経膠腫を有するNF1患者の多くが6歳までに視力低下や眼球突出,斜視を発症するが,小児期後期もしくは成人期まで症状を呈さないこともある[Friedrich & Nuding 2016, Kinore et al 2021].
NF1の小児患者に対する脳のMRI検査で視神経の蛇行が確認されることがあるが,視神経の蛇行とNF1患者における視路神経膠腫の発生に特段の関連はみられない[Ji et al 2013].
その他の腫瘍
神経線維腫
神経線維腫は良性のシュワン細胞の腫瘍で,実質的には全身のあらゆる神経に生じる可能性がある [Brena et al 2020,Serra et al 2020,Ozarslan et al 2021].皮膚の神経線維腫は散在性の境界明瞭な腫瘤で,通常1-2mmから数cmの大きさである.硬さは軟らかいものからゴム状のもの,硬いものまでさまざまである.無茎性のことも有茎性のこともあり,皮膚病変の色調は,隣接する健常皮膚と同じか,もしくはややピンクや褐色,青みのかった色合いを呈する.多くは無症状だが,痒みや圧痛を伴うこともある.皮膚の神経線維腫は小児期にみられることは稀だが,成人のNF1患者にはほぼ例外なくみられる.
皮下の神経線維腫は皮膚よりも下の部分にできる(その上の皮膚は可動性をもつ).多くはゴム状で結節性であるが,中にはびまん性で境界が不明瞭なものや,軟らかいもの,硬さが不均質なものもみられる.表在性のびまん性神経線維腫直上の皮膚については,色素沈着や毛髪の異常がみられることがある.皮下の神経線維腫は孤立性であったり,多数集まって存在していたり,神経の走行に沿って数珠状に連続していることもある.皮下の神経線維腫の大多数は小さいものの,中には直径5cm以上の大きさに達することもある.圧痛があったり,時に強い痛みを伴ったりすることもある.皮下の神経線維腫は小児期にはあまりみられないが,身体診察をおこなうと成人のNF1患者では約15%にみられる.
皮膚や皮下の神経線維腫は生涯にわたって出現しつづけるが,出現の速度は年によって大きなばらつきがある.成人NF1患者の身体診察でみられる神経線維腫の数は,数個から数百,数千個にいたるまで幅がある.女性患者では妊娠中に神経線維腫の数や大きさの急速な増大を経験することがあるが,妊娠経験のない生殖年齢の女性と比べて,腫瘍量の継続的な増大をもたらす訳ではないとされている[Well et al 2020]
叢状神経線維腫
NF1患者の約半数に叢状神経線維腫がみられる.腫瘍の多くは内在性のため,身体診察では明らかにならない.しかし,MRI検査では確認可能である(「画像診断」の項を参照).叢状神経線維腫は小児期から思春期にかけて増大する傾向があるが,その後は成人期を通して安定した状態を保つ[Nguyen et al 2012].叢状神経線維腫の多くは無症状であるものの,痛みの原因となることがあり,増大によって変形が生じることや,隣接組織の異常増殖やびらんを引き起こすことがあり,神経や他の機能に影響を及ぼすこともある.
表在性のびまん性叢状神経線維腫は,軟らかく不規則であり,皮膚病変の肥厚や肥大,色素沈着をきたすことが多い.より広範なびまん性叢状神経線維腫になると,触診で「bag of worms(虫の入った袋)」に触れたような特徴的な感触を示すことがあるが,これは複数の神経や神経枝の関与を意味している.また,叢状神経線維腫の中には硬く結節性のものもあり,それが単独で存在することもあれば,ある程度の長さをもって繋がっていることもあり,場合によっては神経の全長にわたって連なり,触診で「数珠状」の感触を示すこともある.身体診察でわからないような深部の叢状神経線維腫にはびまん性と結節性があり,どの神経や神経根,神経叢においても単発あるいは集合性に生じる可能性がある.
悪性末梢神経鞘腫瘍(MPNST)
悪性末梢神経鞘腫瘍(MPNST)はNF1に伴う最も頻度の高い悪性腫瘍である.NF1患者のMPNSTは一般集団より若い年齢で発症し,予後不良となる傾向がある[Martin et al 2020,Sharma et al 2021].すべてではないにせよほとんどのMPNSTは,すでに存在するびまん性叢状神経線維腫や結節性叢状神経線維腫から発生する.悪性化の徴候として最も多くみられるものは,持続性の疼痛で,新たな症状として発生することもあれば,すでにあった痛みが悪化するといったこともある.臨床的あるいはMRI上でみられるMPNSTの急激な増大ないし性状の変化に伴って,疼痛が生じることもある.
NF1患者の中でもNF1遺伝子の全欠失例や,良性の皮下神経線維腫を有する例,深部に大きな叢状神経線維腫を有する例については,これらの特徴を有しない患者に比べ,MPNSTを発症するリスクが高い[Nguyen et al 2014].非定型的な神経線維腫(atypical neurofibroma)を有する例についても,MPNSTを発生するリスクが非常に高い[Higham et al 2018]
脳腫瘍
NF1患者における視路以外の神経膠腫は通常無症状なため,定期的なスクリーニングとしての頭部MRI検査や,他の症候に対しておこなわれた頭部MRI検査で,偶発的に見つかることがほとんどである.通常は低悪性度の腫瘍であり,長年にわたって全く増大しないか,あるいは増大するにしてもゆっくりである[Sellmer et al 2017].しかし, 症候性や高悪性度の脳腫瘍も時折みられる[Byrne et al 2017,Glombova et al 2019].
視神経以外の神経膠腫を有するNF1患者の少なくとも20%は,複数の病変を有している[Sellmer et al 2017,Glombova et al 2019].視路神経膠腫を有するNF1患者の17〜20%に,新たな中枢神経系神経膠腫(CNS)が生じる[Sharif et al 2006, Sellmer et al 2018]
乳癌
女性NF1患者は,50歳までに乳癌を発症するリスクや[Uusitalo et al 2016],乳癌による死亡リスクがかなり高い[Evans et al 2020].また対側乳癌の累計発症リスクも通常より高い[Evans et al 2020].女性NF1患者においてはHER2陽性乳癌が多く,他の腫瘍マーカーも陽性であることが多い[Evans et al 2020].
血液悪性腫瘍
小児NF1患者において若年性骨髄単球性白血病(JMML)は稀ではあるものの,小児一般集団に比べて数百倍の頻度でみられる[Niemeyer & Flotho 2019].臨床的特徴として,脾腫大や肝腫大,白血病の肺浸潤がみられ,末梢血塗抹標本では骨髄球や後骨髄球,そして時には有核赤血球が認められる.JMMLを伴う小児NF1患者に若年性黄色肉芽腫がみられることがあるものの,それ以外の小児NF1患者に比べて多いというわけではない[Liy-Wong et al, 2017].成人NF1患者においてリンパ網内系の悪性腫瘍の頻度が高いかどうかは不明である[Bergqvistら2021].
その他の腫瘍.
NF1患者においては,横紋筋肉腫[Crucis et al 2015]や褐色細胞腫[Gruber et al 2017],パラガングリオーマ[Gruber et al 2017],消化管間質腫瘍[Nishida et al 2016],グロムス腫瘍[Kumar et al 2014] など様々な腫瘍が通常より頻繁にみられることがある.また,NF1患者はその他のがんの発症リスクも高い可能性がある[Seminog & Goldacre 2013, Varan et al 2016,Landry et al 2021].
その他の神経学的症候
運動機能.
小児NF1患者では,筋緊張低下や協調運動障害,平衡障害,巧緻運動障害がしばしばみられる[Iannuzzi et al 2016,Haas-Lude et al 2018,Pardej et al 2022].小児NF1患者は,同性・同年齢で体重も同程度の非罹患児と比べると,筋力も弱い[Summers et al 2015].
知的障害と学習障害.
NF1患者においては視空間認知障害が最もよくみられるが,限局性学習症や実行機能障害,記憶障害や言語障害も頻繁に認められる[Vogel et al 2017].NF1患者の平均IQは一般集団よりも最大で1SD低く,知的障害といえるレベルの低下(IQ<70)はNF1患者の4%-8%にみられるが,これは一般集団における頻度の2倍にあたる[Vogel et al 2017,Al-Farsi et al 2022].知的障害はNF1遺伝子の全欠失を有する患者においてより多く認められる.
行動上の問題.
行動上の問題としては,社会的能力や注意力に関する問題が挙げられる.NF1患者では小児と成人のいずれにおいても,孤立や周囲の理解を得にくいといった社会的困難が増し,社会的スキルの低さや向社会的行動をとりにくいことが報告されている[Chisholm et al 2018,Payne et al 2020]. 注意欠陥多動性障害は小児や思春期のNF1患者の30%-50%にみられ[Voge et al 2017,Chisholm et al 2018,Domon-Archambault et al 2018],これが成人期まで存続することもある[Mautneret al 2015]. 小児NF1患者ではしばしば自閉的傾向がみられ,小児NF1患者の25%が自閉スペクトラム症の標準的な診断基準を満たす[Vogel et al 2017,Chisholm et al 2018,Domon-Archambault et al 2018,Payne et al 2020,Chisholm et al 2022].成人NF1患者における自閉的傾向は,小児ほどは多くないとされている[Morris et al 2016].年齢を問わず,NF1患者には睡眠障害が多くみられる[Domon-Archambault et al 2018,Fjermestad et al 2018].成人NF1患者では,気分障害,不安障害,境界性パーソナリティ障害などの精神疾患が通常よりも多くみられる[Domon-Archambault et al 2018,Kenborg et al 2021].
多発神経障害.
びまん性の多発神経障害はNF1患者の数パーセントにみられ,全例ではないもののその多くは多発性の神経根腫瘍を有する[Barnett et al 2019,Bayat & Bayat 2020].NF1の多発神経障害は無症状の場合もあれば,感覚障害や疼痛,痒みを伴うこともある.MPNSTのリスクは,多発神経障害を有する患者では有しない患者より高いとされている.
てんかん.
てんかん発作はNF1患者の約5%に認められ,小児よりも成人にやや多くみられる[Bernardo et al 2020,Sorrentino et al 2021].全般発作のこともあるが多くは焦点発作で,脳腫瘍や梗塞部位,内側側頭葉硬化に伴って生じる[Pecoraro et al 2017,Bernardo et al 2020,Sorrentino et al 2021].神経発達障害は,てんかんを有するNF1患者により多くみられる[Sorrentino et al 2021].
NF1患者におけるてんかんの治療法は,NF1でない一般の例と変わるところがない[Bernardo et al 2020,Sorrentino et al 2021].焦点発作のコントロールには,複数の抗てんかん薬の使用,または脳の病変部位に対する外科的切除が必要となることがある.
頭痛.
頭痛はNF1患者の半数以上に認められ,小児よりも成人に多くみられる[Fjermestad et al 2018,Hirabaru & Matsuo 2018,Kongkriangkai et al 2019].NF1患者では片頭痛が最もよくみられるものの,他の種類の頭痛が生じることもある.頭痛がある患者の頭部MRI検査でNF1関連病変が確認されることがよくあるが,頻度的には頭痛のない患者と大差ない[Afridi et al 2015].
神経画像.
脳のMRI検査のT2強調画像では,小児NF1患者の半数以上に高信号域(unidentified bright objects 〈UBOs〉 もしくはfocal areas of signal intensity 〈FASI〉)を認める[Sellmer et al 2018].これらの特徴は視路,基底核,脳幹,小脳,大脳皮質にみられるものの,通常占拠性病変の所見は認められない.典型的なUBOsはT1強調画像やCT検査では捉えられない.UBOsは病理学的には海綿状の髄鞘障害の領域に相当している[DiPaolo et al 1995].数や大きさの点でのUBOsの出現ピークはおおむね7歳で,その後は消退傾向を示すものの,成人期まで残存することもある[Sellmer et al 2018,Calvez et al 2020].UBOsの存在は小児NF1患者のてんかん発作とは関連しない[Hsieh et al 2011].経時的に見たときのUBOsの存在,数,大きさ,位置,あるいは消失が,小児NF1患者の学習障害や行動障害と関連しているという研究が複数あるが,研究者間で意見の一致には至っていない[Payne et al 2014, Roy et al 2015,Parmeggiani et al 2018,Eby et al 2019,Baudou et al 2020].
脳梁の拡張は一部の小児NF1患者にみられ,学習障害と関連している[Pride et al 2010, Aydin et al 2016].
MRI検査は,視路神経膠腫や脳腫瘍,脳神経・脊髄神経・末梢神経の神経線維腫,全身に生じうるびまん性ないし結節性の神経線維腫といったものを発見する上で有用な画像診断法である.陽電子放出断層撮影(PET検査)は,MPNSTを把握する上で有用であり[Nishida et al 2021],高解像度超音波検査は,皮膚ならびに表在性の叢状神経線維腫の特徴を明らかにする上で有用である[Winter et al 2020].
筋骨格の特徴
長管骨,蝶形骨翼,椎体の異形成
NF1患者における骨の異形成は,原発性に生じること(主に長管骨の異形成)もあれば,叢状神経線維腫の隣接や硬膜拡張に伴って起こること(椎体や蝶形骨翼の異形成)もある.これらの限局性病変における骨折または骨の欠損は,治療に難渋することが多い[Elefteriou et al 2009],長管骨や頭蓋顔面骨,脊椎の異形成とそれに伴う変形に対する外科的治療は困難を伴うことが多いため,経験豊富な専門医によりおこなうのが最善である[Mladenov et al 2020].
長管骨の異形成
長管骨(多くの場合脛骨や腓骨)の異形成は稀であるもののNF1特有の徴候であり[Elefteriou et al 2009],先天性でほとんどの場合片側性である.通常,乳児期に下腿が前外側へ弯曲しているが,早期発見ができれば,装具の使用により骨折を防げる可能性がある.初期の画像所見として,弯曲頂部の皮質肥厚による髄腔狭小化がみられる[Stevenson et al 2007].
蝶形骨翼の異形成
蝶形骨翼の異形成は眼窩が非対称になるのが一般的だが,頭蓋の画像検査で偶然見つかることもある.蝶形骨翼の異形成は大抵の場合は進行しないが,時に進行すると眼窩が破壊され,拍動性眼球陥凹を引き起こす[Chauvel-Picard et al 2020].
脊椎の異形成
脊椎の異形成は通常dystrophic typeの側彎症として,一般的な思春期側彎症よりもかなり早く,6歳〜8歳で出現することが多い.dystrophic typeの側彎症は,短い分節で高度な弯曲を呈することを特徴とし,発見後数か月のうちに急速に進行することがある[Kaspiris et al 2022].
non-dystrophic typeの側彎症
NF1患者にみられるnon-dystrophic typeの側彎症は脊椎の異常を伴わないことが多く,発症年齢や良性の経過をたどるという点で一般的な思春期側彎症と類似している[Elefteriou et al 2009].
骨粗鬆症
NF1患者では,全身の骨量減少が通常よりも高頻度にみられ[Rodari et al 2018,Filopanti et al 2019,Jalabert et al 2021,Kaspiris et al 2022],骨折も通常より多く発生する[Heervä et al 2012].成人NF1患者では骨粗鬆症を一般集団より高頻度かつ早期に発症する[Filopanti et al 2019,Kaspiris et al 2022].これら骨性変化の原因は明らかでないが,NF1患者では血清25-ヒドロキシビタミンD濃度が基準値を下回ることや,副甲状腺ホルモン値の上昇,骨吸収の亢進がよくみられる [Riccardi et al 2020,Tezol et al 2021,Kaspiris et al 2022].NF1患者では骨芽細胞や破骨細胞の機能が正常ではないと考えられる[Riccardi et al 2020,Kaspiris et al 2022].
血管病変
血管性高血圧
NF1患者の少なくとも15%-20%に血管性高血圧がみられる[Dubov et al 2016,Sivasubramanian & Meyers 2021].これは,いずれの年齢にも生じうるが,小児よりも成人に多くみられる.特定の原因が見出されないことが多いものの,特に小児においては,NF1に特徴的な血管病変である腎動脈狭窄や腹部大動脈縮窄症が高血圧の原因となっていることがある[Celik et al 2021,Sivasubramanian & Meyers 2021].NF1関連高血圧症の例において水腎症やその他の尿路の器質的異常が多くみられはするものの,高血圧を有しない例においてもみられることがある[Dubov et al 2016,Celik et al 2021].
褐色細胞腫やパラガングリオーマは一般集団に比較してNF1患者に多いとされるが,成人NF1患者にみられるのは1%未満である.これらの腫瘍は無症状であることが多いものの,動脈性高血圧を引き起こす可能性がある[Al-Sharefi et al 2019].
肺高血圧症
稀ではあるものの,肺高血圧症は高齢のNF1患者に生じうる非常に重大な合併症である[Jutant et al 2018,Jutant et al 2020].肺高血圧症は肺実質病変を伴うことが多く,それ自体が肺の血管に影響を及ぼすNF1の血管障害の一徴候であるとも考えられる[Jutant et al 2018].
脳卒中
NF1患者では脳卒中の頻度が高く,一般集団に比べて若年で発症することが多い[Terry et al 2016]. 解剖学的に異形な大脳動脈の狭窄や拡張,頭蓋内動脈瘤は,一般集団に比べてNF1患者に高頻度に認められる[Bekiesińska-Figatowska et al 2014, D'Arco et al 2014,Barreto-Duarte et al 2021].問題になる部位としては,内頚動脈,中大脳動脈,前大脳動脈が最も多い.狭窄領域周辺では小さな拡張毛細血管が形成され,脳血管造影で立ちのぼる煙のような像(もやもや)を呈する.原発性脳腫瘍に対して頭部への放射線照射をおこなった小児NF1患者は,もやもや血管が生じる可能性が3倍になる[ Murphy et al 2015].
心臓の課題
フィンランドの複数の集団ベースレジストリに基づく一研究によると,小児NF1患者における循環器系の先天異常の発生頻度は,通常の3.35倍(95%の信頼区間で1.64-6.83倍)に達するという[Leppävirta et al 2018].NF1患者に最も多くみられる心奇形は,肺動脈弁狭窄および僧帽弁の奇形である.[Lin et al 2000, Pinna et al 2019].先天性心疾患または肥大型心筋症は,NF1遺伝子の全欠失を有する患者に特によくみられる[Nguyen et al 2013, Pinna et al 2019].心臓内に神経線維腫が生じることもある[Nguyen et al 2013].
肺疾患
NF1関連のびまん性肺疾患は,成人NF1患者の10%-20%にみられる[Jutant et al 2018,Alves Júnior et al 2019]. 症状は非特異的なことが多く,労作性呼吸困難,息切れ,慢性咳嗽,胸痛などがある.初めて症状が出現するのはふつう30歳代ないし40歳代であるが,特有の徴候は小児NF1患者の画像解析でもみられることがある[Spinnato et al 2019].NF1関連のびまん性肺疾患というと,通常は上葉に生じる嚢胞性や水泡性の肺疾患と,肺底部の間質性肺疾患であり,それらの同定には胸部CT検査が用いられる.NF1関連肺疾患は,往々にして既存の治療法に対する反応性が低い.
成長発達
NF1患者の身長は平均を下回り,頭囲は平均を上回る傾向がある[Zessis et al 2018].しかし,身長が-3SDを下回ることや,頭囲が+4SDを上回ることは非常に稀である.
一方,NF1遺伝子の全欠失を有する患者では, 2~6歳にかけて過成長(特に身長)を呈する[Ning et al 2016,Kehrer-Sawatzki et al 2020].こうした一部の小児患者の臨床像はWeaver症候群との類似性がみられる.(「遺伝子型-表現型相関」の項を参照)
男女とも性成熟の速度は遅いものの,二次性徴は通常正常である[Zessis et al 2018].ただし,思春期発来の遅れがよく見られ[Virdis et al 2003],特に視交叉に腫瘍を有する小児NF1患者では,思春期早発症ないしは成長ホルモン分泌亢進症がみられることもある[Cambiaso et al 2017,Hannah-Shmouni & Stratakis 2019].
寿命
NF1患者の寿命の中央値は一般集団より8年以上短い[Evans et al 2011, Wilding et al 2012].女性の寿命はさらに短くなるとの指摘もある[Uusitalo et al 2015].悪性腫瘍(MPNST)や血管病変は,NF1患者における最も重要な早期の死因となっている[Evans et al 2011, Masocco et al 2011,Uusitalo et al 2015].
生活の質(QOL)
小児NF1患者と成人NF1患者の両方において,生活の質(QOL)の評価は対照群よりも低い[Vranceanu et al 2015].整容面,医療面,社会面,行動面に関するNF1の特徴は,いずれもNF1患者の生活の質を低下させる可能性があり,抑うつにより本来有している能力をうまく発揮できない状況に至ることもある[Domon-Archambault et al 2018].デンマークにおける集団ベースのレジストリ研究によると,NF1患者は一般集団に比べ,すべての年齢層において入院の経験が2倍以上あり,頻回で入院期間も長いことが示された[Kenborg et al 2020].
NF1の多様な表現型
NF1モザイク
NF1モザイク(NF1遺伝子病的バリアントの体細胞モザイク)では,NF1の臨床症候が身体の一部ないしは複数の部位に限局することもあれば,典型的なNF1と同じように全身性に生じることもある[Ejerskov et al 2021].NF1モザイクは同じ病的バリアントを有する場合であっても典型的なNF1に比べて軽症であり, NF1モザイクの成人の中にはNF1の臨床的特徴が全くみられない例もある[Kluwe et al 2020,Yang et al 2020].身体の一部分のみに症状を呈するNF1モザイクはしばしば「segmental NF1(分節型NF1)」と呼ばれるが,特に幼児期ではNF1モザイクではない症例であっても,偶然身体の一部にしか所見がみられていないこともあるため,実情により近い表現として「localized mosaic NF1(限局性NF1モザイク)」という用語がよく用いられる.成人のNF1モザイクの子どもが典型的な(非モザイクの)NF1となることもある[Legius & Brems 2020](「遺伝カウンセリング」の項を参照).
NF1-Noonan症候群
NF1-Noonan症候群の表現型はNF1患者の約12%に見られる.臨床所見としては眼間開離,眼瞼裂斜下,眼瞼下垂,耳介低位,翼状頚,胸郭変形,肺動脈弁狭窄がある.これらの所見を呈するNF1患者の血縁者は,Noonan症候群の所見を伴うこともあれば,伴わないこともある[Chen et al 2014,Ekvall et al 2014].NF1-Noonan症候群の表現型には遺伝的異質性がみられる.NF1-Noonan症候群患者の中にはNF1遺伝子とPTPN11遺伝子(Noonan症候群の原因遺伝子として最も一般的なもの)の両方に病的バリアントが認められる例がある[D'Amicoet al 2021].一方で,病的バリアントがNF1遺伝子にしか確認されないような例もみられる.
Watson症候群は,NF1と重なる表現型として肺動脈弁狭窄や,カフェ・オ・レ斑,低身長,軽度の知的障害といった特徴を有し,NF1遺伝子の病的バリアントに起因する[Allanson et al1991].
家族性脊髄神経線維腫
家族性脊髄神経線維腫はあらゆる神経根に神経線維腫を生じるもののNF1の皮膚所見はほとんどみられない[Bettegowda et al 2021].その疾患名とは裏腹に 孤発例も散見される[Ruggieri et al 2015].
多発性脊髄神経節神経腫
多発性の脊髄神経節神経腫(神経線維腫ではない)および多発性の皮下腫瘍については,臨床症候ではNF1診断基準を満たさないもののNF1遺伝子の病的バリアントを有する成人の1例が報告されている[Bacci et al 2010].
多発性脂肪腫
NF1遺伝子の病的バリアントを有し,多発性脂肪腫がみられるものの,他にNF1の症候が全くみられない例が報告されている[Ramirez et al 2021].
視神経膠腫
症候性の視神経膠腫以外にはNF1の診断的特徴がみられないものの,NF1遺伝子の病的バリアントを有する21歳男性の例が報告されている[Buske et al 1999].
脳頭蓋皮膚脂肪腫症
脳頭蓋皮膚脂肪腫症は,5個以上のカフェ・オ・レ斑とNF1遺伝子の病的バリアントを有する小児において一例報告されている[Legius et al 1995].
NF1においては遺伝子型と表現型の相関が複数報告されている.
- 1.4MbにわたるNF1遺伝子の全欠失(タイプ1)を有する患者では,皮膚の神経線維腫や叢状神経線維腫が早期に多数出現し,MPNSTの発症リスクが高く,認知機能障害もより高頻度かつ重度で,身体の過剰発育や大きな手足が認められる [Bettegowda et al 2021,Kehrer-Sawatzki & Cooper 2021,Pacot et al 2021,Well et al 2021].頻繁にみられる形態的な特徴として,粗野な顔貌,平坦な前額,眼間開離,広い鼻尖,耳介低位,幅広の頸が,思春期や成人期にしばしば認められる[Kehrer-Sawatzki & Cooper 2021].
- コドン844から848[NM_000267.3]までの5つコドン(この部分はニューロフィブロミン中のシステイン‐セリンリッチドメインをコードする)のいずれかにミスセンスバリアントを有する成人については,叢状神経線維腫ないし脊髄神経線維腫,視路神経膠腫,悪性新生物,骨格異常などが頻発し,まれに重度の表現型を呈することが報告されている[Koczkowska et al 2018,Bettegowda et al 2021].
- p.Met992delについては,NF1にみられる典型的な色素斑の症候を伴うが,皮膚,皮下,ないし体表の叢状神経線維腫は現れない[Bettegowda et al 2021].このバリアントを有する患者の4分の1はNF1の臨床診断基準を満たさないものの,視路以外の脳腫瘍,認知機能障害や学習障害,Noonan症候群の症候に関しては他のNF1患者と同様の頻度で出現する.
- p.Arg1038Glyについては,NF1の症候のうち色素斑を軽度に有するものの神経線維腫は少なく,NF1-Noonan症候群の症候を示す例が多くみられる[Trevisson et al 2019].
- p.Met1149のミスセンスバリアントについては,色素症状,学習障害の頻発,NF1-Noonan症候群の症候といった,比較的軽度の表現型を示す[Koczkowska et al 2020].
- p.Arg1276のミスセンスバリアントについては,肺動脈弁狭窄に代表される心血管奇形やNF1-Noonan症候群の表現型や症候性脊髄神経線維腫が通常より高頻度で生じる[Koczkowska et al 2020].
- p.Lys1423のミスセンスバリアントについては,叢状神経線維腫,学習障害,心血管系奇形(特に肺動脈弁狭窄),NF1-Noonan症候群の表現型が通常より高頻度で生じる[Koczkowska et al 2020].
- p.Arg1809のミスセンスバリアントのうちの一部は,皮膚の神経線維腫や臨床的に明らかな叢状神経線維腫は生じないものの多発性のカフェ・オ・レ斑を伴い,学習障害,低身長,肺動脈弁狭窄もしばしばみられる [Pinna et al 2015,Rojnueangnit et al 2015,Bettegowda et al 2021].
浸透率
家系の調査によると,NF1の浸透率は小児期を過ぎるとほぼ100%となる[Rasmussen & Friedman 2000],ただし,分子遺伝学的検査では,NF1の病的バリアントによる不完全浸透の例がわずかだが報告されている[Bettegowda et al 2021].
病名
NF1でも中枢神経病変を生じるものの,NF2(神経線維腫症2型・中枢神経線維腫症)と区別するために「末梢神経線維腫症」と呼ばれていた.
NF1を指す際に,特に何の限定もなく「神経線維腫症」という名称が文献中に用いられることがあるが,他の著者がNF1に加えてLegius症候群やNF2,シュワノマトーシスを包括する一群を指す用語として「神経線維腫症」を用いることがあるため混乱が生じる.
頻度
フィンランドの全国民を対象とした登録ベースの研究において,NF1の有病率は0歳から74歳までの人の2,052人に1人の割合と推定されている[Kallionpää et al 2018].有病率は,最も若いコホートで最も高く,年齢の高いコホートになると低くなっており,これは死亡率の状況を反映したものである(「寿命」の項を参照).この研究によると,NF1の出生頻度は1,871人に1人と推定されている[Uusitalo et al 2015]ことから,NF1は常染色体顕性遺伝(優性遺伝)性疾患の中で最も頻度の高い疾患のひとつとなっている.この研究における有病率ならびに出生頻度の値は,それ以前の研究[Evansら2010]に比べて高かったが,これは,両集団間に実際に存在した出生頻度・有病率の違いというよりも,表現型がより軽度な患者を含めて集計した結果だと思われる.
患者の約半数は新生(de novo)の病的バリアントにより発症する.NF1の出生頻度に関するフィンランドの研究の推定値が正しい場合で,患者の妊孕性が通常の半分に低下している[Huson et al 1989]と仮定すると,NF1遺伝子の変異発生率は1/8,000を超えることになり,これまでに知られているヒトの遺伝子の中では最も高いことになる.このように変異率が異常に高くなっている原因ははまだ明らかでない.
遺伝学的に関連のある疾患(同一アレル疾患)
NF1遺伝子の生殖細胞系列の病的バリアントに関連する疾患は,このGeneReviewで述べたもの以外は知られていない.
鑑別診断
カフェ・オ・レ斑(CALMs)や神経線維腫症1型(NF1)でみられる他の症候を有する遺伝性疾患や先天異常症候群は100種以上知られているが,NF1との鑑別が困難なものはわずかしかない.NF1との鑑別診断において検討すべき疾患を表3にまとめて示した.
カフェ・オ・レ斑(CALMs)や神経線維腫症1型(NF1)でみられる他の症候を有する遺伝性疾患や先天異常症候群は100種以上知られているが,NF1との鑑別が困難なものはわずかしかない.NF1との鑑別診断において検討すべき疾患を表3にまとめて示した.
表3:神経線維腫症1型との鑑別診断において着目する遺伝子
| 遺伝子 | 疾患名 | 遺伝形式 | 臨床的特徴/コメント |
|---|---|---|---|
| AKT1 1 | Proteus症候群 | 脚注1参照 | 結合織母斑や表皮母斑,その他組織の過成長,および骨化過剰 |
| BRAF MAP2K1 PTPN11 RAF1 |
多発性黒子を伴う Noonan症候群(以前はLEOPARD症候群と呼ばれていた) |
AD | 多発性黒子,眼間開離,難聴,先天性心疾患 |
| BRAF KRAS LZTR1 MAP2K1 NRAS PTPN11 RAF1 RIT1 SOS1 |
Noonan症候群(NS) | AD (AR)2 |
低身長,先天性心疾患,翼状頸,特徴的顔貌. NS様の顔貌の特徴がNF1患者にみられることもある. NSの顔貌の特徴は年齢とともに変化する. 年齢に関係なくみられる特徴:厚みのある耳輪で位置が低く後方に傾斜した耳介,鮮やかな青色または青緑色の虹彩,眼間開離,眼瞼裂斜下,内眼角贅皮,眼瞼下垂 |
| GNAS 3 | 線維性骨異形成症/McCune-Albright症候群(FD/MAS) | 脚注3参照 | 辺縁不整の大きなCALMs,多発性線維性骨異形成症. |
| KIT SNAI2 |
限局性白皮症 (OMIM 172800) |
AD | 皮膚に色素沈着と色素脱失がみられ,色素脱失領域を色素沈着過剰な領域が縁取る.前髪の脱色 |
| LZTR1 SMARCB1 |
シュワノマトーシス | AD | 多発性の神経鞘腫や髄膜腫(後者は低頻度)発生の素因を有する 最も一般的な所見:限局性またはびまん性の疼痛,あるいは無症候性の腫瘤. |
| MLH1 MSH2 MSH6 PMS2 |
先天性ミスマッチ修復遺伝子異常症 (CMMRD;「Lynch症候群」を参照) |
AR | 稀な小児がん素因の症候群.20歳未満で大腸癌や小腸癌を発生しやすい. 皮膚の表現型は NF1 に酷似している. CMMRDは,両親に近親婚が多く,片親もしくは両親において,左記の遺伝子いずれかのヘテロ接合性病的バリアントに起因するLynch症候群の臨床症候や家族歴を有することが多い点で,NF1と区別することができる. 一般的に両親いずれもNF1にみられる臨床症候は有しない. |
| NF2 | 神経線維腫症2型(NF2) | AD | 両側性前庭経鞘腫,脳神経・末梢神経の神経鞘腫,皮膚の神経鞘腫,髄膜腫,若年性後嚢下白内障 |
| PDGFRB | 乳児筋線維腫症(OMIM 228550) | AD | 皮膚,皮下組織,骨格筋,骨,内臓の多発性腫瘍 |
| SPRED1 | Legius症候群 | AD | 神経線維腫やその他の腫瘍,虹彩小結節を伴わない多発CALMs. |
AD=常染色体顕性遺伝(優性遺伝),AR=常染色体潜性遺伝(劣性遺伝),CALMs=カフェ・オ・レ斑,NS=Nooonan症候群
- Proteus症候群の診断基準を満たす患者の90%以上に,AKT1遺伝子の病的バリアントの体細胞モザイクが同定されている.
世代間の継承例や同胞の発症例は確認されていない. - NSの大多数は常染色体顕性遺伝だが,LZTR1遺伝子の病的バリアントについては,常染色体顕性遺伝と常染色体潜性遺伝のいずれの場合もある.
- FD/MASは,初期胚発生後に体細胞性に生じたGNAS遺伝子の活性型バリアントによる.FD/MASの世代間の継承例は確認されていない.
(訳注:「4」は,表中にも脚注にもない) - NF1患者に神経線維腫や虹彩小結節がみられるのは通常小児期後期や思春期のため,幼少期にはLegius症候群とNF1を臨床的に鑑別できないことがある.両親がLegius症候群やNF1の症候を有しているか診査することで両者を鑑別できる場合もあるが,孤発例については思春期以降に再評価を行うか,あるいは分子遺伝学的検査により診断を確定することが必要な場合もある[Legiu et al 2021].
NF1の特徴を有するその他の疾患
- 孤立性の家族性多発性カフェ・オ・レ斑(OMIM 114030)
家族性多発性にCALMsを有するもののNF1のその他の臨床症候を有しない253例では,86.6%にNF1遺伝子の病的バリアントが,7.1%にSPRED1遺伝子の病的バリアントが認められたものの,6.3%はNF1遺伝子にもSPRED1遺伝子にも病的バリアントが同定されなかった[Messiaen et al 2009].
- 多発性眼窩神経線維腫,痛みを伴う末梢神経腫瘍,特徴的顔貌,Marfan様体型[Babovic-Vuksanovic et al 2012]
臨床的マネジメント
米国小児学会と米国臨床遺伝・ゲノム学会(American College of Medical Genetics and Genomics:ACMG)は小児NF1患者に対する治療ガイドラインを公表しており[Miller et al 2019],ACMGは成人NF1患者に対する治療ガイドラインも公表している[Stewart et al 2018].
NFフランスネットワーク[Bergqvist et al 2020]や他の専門家集団も同様の提言を発表している[Ly & Blakeley 2019,Baudou & Chaix 2020]. NF1の豊富な治療経験と高い関心を有する幅広い分野の専門医が在籍するNFクリニックに紹介することは,多くの患者にとって有益であろうと思われる[Toledano-Alhadef et al 2020].
最初の診断に続いて行う評価
神経線維腫症1型(NF1)と診断された患者の臨床像やニーズを把握するために,表4に示す評価項目の実施が(診断時の評価の一部として実施されていない場合には)推奨されている.
表4:診断後の神経線維腫症1型患者に対して推奨される評価
| 系/懸念事項 | 評価項目 | コメント |
|---|---|---|
| 皮膚 | 神経線維腫および叢状神経線維腫に関する皮膚の臨床評価 | |
| 眼 | 眼底検査,虹彩の細隙灯顕微鏡検査,眼底の近赤外線反射イメージングまたは光干渉断層画像化法(OCT)などの眼科的評価,および視力評価 | |
| 神経 | 神経学的検査;てんかん,頭痛,疼痛に関する評価 | 注:症状のない患者に対する定期的な脳MRI検査実施については意見が分かれる1. |
| 発達 | 発達評価 | |
| 精神 | 精神神経学的評価 | |
| 骨格 | 非対称性,長管骨異形成,蝶形骨翼異形成,脊椎異形成の臨床評価,および側彎症や頻回な骨折 | |
| 血管 | 血圧評価 | 高血圧を有する患者については,腎血管疾患,腹部大動脈縮窄症,水腎症、その他の尿路の器質的異常,褐色細胞腫やパラガングリオーマの評価を行う[Sivasubramanian & Meyers 2021]. |
| 心臓 | 先天性心疾患または心筋症の徴候や症状に関する病歴聴取と臨床検査 | |
| 成長 | 成長曲線シートに身長,体重,頭囲の値をプロットする. | |
| 遺伝カウンセリング | 遺伝の専門家2がおこなう | 患者および家族に対してNF1の自然歴や遺伝形式,およびその影響について情報提供を行い,医療上あるいは個人に関する意思決定を支援する |
- 提唱者は脳MRI検査の実施によって臨床症候が出現する前に脳頭蓋骨の形態異常や腫瘍,あるいは血管病変を検出することができるため有用であると主張している.一方で無症状の患者に対する頭部MRI検査に反対する者は,unidentified bright objects(UBOs)の臨床的意義が不明であることや,費用がかさむこと,幼児では鎮静が必要となること,何らかの所見がみられた場合には確認のために繰り返し検査が必要になることを指摘している.
- 臨床遺伝専門医,認定遺伝カウンセラー,遺伝看護専門看護師
症候に対する治療
表5:神経線維腫症1型患者の症候に対する治療
| 症候/懸念事項 | 治療 | 考慮事項/その他 |
|---|---|---|
視路神経膠腫 |
|
|
皮膚/皮下の神経線維腫 |
美容上問題となる場合や不快感を伴う部位(例えばベルトや襟が当たる部位)に生じた神経線維腫は,外科的切除やレーザー,電気焼灼を行う. |
レーザー治療は,多数の神経線維腫を迅速かつ効果的に除去でき,美容面でも満足のいく結果が得られる[Méni et al 2015]. |
叢状神経線維腫 |
訳注:本邦においては2022年より小児に関して、NF1に伴う手術不能な叢状神経線維腫に対する治療薬として,MEK阻害剤セルメチニブが保険収載されている |
|
悪性末梢神経鞘腫瘍(MPNST) |
|
MEK阻害剤や免疫療法,放射線治療について,現在臨床試験が行われている段階である[Marjanska et al 2020]. |
脳腫瘍 |
脳MRI検査を用いた観察,ならびにNF1に精通した腫瘍専門医による管理 |
|
乳癌 |
NF1患者では乳癌の悪性度が高いとする報告があるものの,NF1女性患者についてもNF1でない女性と同様に病理所見や腫瘍マーカーを基準とした治療を行う. |
可能であれば,放射線治療は避けるのが適切である. |
造血器腫瘍 |
NF1の治療に精通した腫瘍専門医による管理 |
|
疼痛 |
|
従来の治療法をおこなってもADLが低下するような難治性の疼痛を示す例については,疼痛に詳しい専門医への紹介が必要である. |
発達遅滞/知的障害 |
発達に関する専門医による管理[Miller et al 2019] |
|
行動/精神症状 |
注意欠陥多動性障害の小児にはメチルフェニデートによる治療が有効なことがある[Lion-François et al 2014]. |
|
てんかん |
てんかん治療に経験豊富な神経内科医による管理 |
|
頭痛 |
頭痛治療に経験豊富な神経内科医による管理 |
|
長管骨異形成 |
|
脛骨の偽関節に対する外科的治療は難しく,満足のいく結果が得られないことが多い[Paley 2019]. |
当該部の叢状神経線維腫を伴う,あるいは伴わない蝶形骨翼異形成 |
NF1治療に経験豊富な頭蓋顔面の専門医チームによる管理 |
小児NF1患者にみられる眼窩/眼窩周囲の叢状神経線維腫に対する臨床管理の提言が, 集学的な専門家集団により作成されている[Avery et al 2017]. |
椎体異形成/dystrophic typeの側彎症 |
NF1治療に経験豊富な整形外科医や脊椎の専門医による管理 |
外科的治療が必要となることが多いものの,手術は複雑かつ困難である[Jia et al 2021]. |
側彎症(non-dystrophic type) |
整形外科医による管理 |
特発性脊柱側彎症に準じた治療 |
骨減少症/頻回の骨折 |
|
年齢を問わず,NF1患者にはビタミンD欠乏症が多くみられる. |
高血圧 |
高血圧の原因に応じて,腎臓内科医ないし循環器内科医による治療を行う[Sivasubramanian & Meyers 2021]. |
|
脳卒中/もやもや病 |
|
|
心疾患 |
循環器内科医や心臓血管外科医による管理 |
|
成長障害/思春期遅発症 |
小児内分泌内科医による管理 |
|
思春期早発症 |
内分泌内科医による管理 |
|
- Gross et al [2020],Galvin at al [2021],Mukhopadhyay et al [2021],Anderson et al [2022]
- 他のMEK阻害剤を用いた叢状神経線維腫の治療に関しては,現在臨床試験による評価が行われているところである[Marjanska et al 2020,Solares t al 2021].
画像診断
MRI検査は,叢状神経線維腫の病変の大きさや広がりの評価[Ahlawat et al 2016],その経時的変化の観察に適している[Nguyen et al 2012].同時に,MRI検査はNF1患者の視路神経膠腫やその他の脳腫瘍,脳の形態異常,脳血管障害の徴候を明らかにするのにも用いられる[Lin et al 2011, Prada et al 2015, Blanchard et al 2016, Sellmer et al 2017, Sellmer et al 2018].また,MRA検査はNF1の血管病変の評価に有用である[D'Arco et al 2014].従来のX線検査でもNF1患者に生じる骨格異常を検出できるが[Patel & Stacy 2012],骨病変の外科的治療が予定されている場合には,CT検査あるいは3D-CT検査が必要となることがある.PET検査やPET-CT検査は末梢神経鞘腫瘍の良悪性の判定に役立つが[Combemale et al 2014, Hirbe &Gutmann 2014, Salamon et al 2014, Chirindel et al 2015, Salamon et al 2015, Van Der Gucht et al 2016],最終的な鑑別は腫瘍の組織学的検査によってのみおこなわれる.PET-CT検査は悪性化が疑われる末梢神経鞘腫瘍の経皮的生検時のガイドとして有用とされている[Brahmi et al 2015].
サーベイランス
米国臨床遺伝・ゲノム学会(ACMG)は小児および成人のNF1患者に対するサーベイランスに関する提言を公表している[Stewart et al 2018,Miller et al 2019].表6a(小児NF1患者に対する定期的なサーベイランス),表6b(NF1成人患者に対する定期的なサーベイランス)にある評価項目は,これらの提言をもとにまとめたものである.
表6a:神経線維腫症1型の小児に対して推奨されるサーベイランス
| 体系/懸念事項 | 評価項目 | 実施頻度 |
|---|---|---|
眼 |
眼科的評価 |
思春期までは年1回,または眼科医の推奨する間隔で実施 |
腫瘍 |
神経線維腫や,新規発生あるいは変化がみられた叢状神経線維腫,その他悪性腫瘍の徴候や症状に対するかかりつけ医による身体診察 |
年1回1 |
神経系 |
神経障害,てんかん発作,頭痛,疼痛に関する神経学的評価 |
年1回1 |
神経発達 |
|
必要に応じて |
骨格系 |
非対称や脊柱側彎に関する臨床的評価 |
小児について成長期が終わるまでの間,年1回1 |
骨折の増加に関する評価 |
年1回 |
|
心血管系 |
|
年1回,ならびに外科的手術の術前1 |
成長障害 |
NF1の成長曲線上における身長,頭囲の評価 |
小児期を通じて年1回 |
内分泌の症候 |
性的成熟の評価 |
小児期早期を通じて年に回 |
- NF1遺伝子全欠失を有する患者,大きいもしくは増大する叢状神経線維腫や頭蓋内腫瘍を有する患者,症候性の血管障害,進行性の骨病変,その他の重大な症候を有する患者については,症状に応じたより頻繁なフォローアップを行う必要がある.
表6b:神経線維腫症1型の成人に関して推奨されているサーベイランス
| 体系/懸念事項 | 評価項目 |
実施頻度 |
|---|---|---|
腫瘍 |
神経線維腫,新規発生ないし変化のみられる叢状神経線維腫,その他,悪性腫瘍の徴候/症状に関し,かかりつけ医による身体診察 |
年1回 |
乳癌 |
マンモグラフィー1 |
30歳以降,年1回 |
乳房造影MRI検査1 |
30歳から50歳の間,年1回実施を考慮 |
|
神経系 |
神経障害,頭痛,てんかん発作,睡眠障害,疼痛に関する神経学的評価 |
年1回 |
精神神経系 |
認知の問題や抑鬱に関する評価 |
年1回,あるいは必要に応じて. |
骨格系 |
脊柱側彎や骨粗鬆症に関する臨床的評価 |
年1回 |
血清ビタミンD値 |
必要に応じて |
|
心血管系 |
|
年に1度. |
- こうしたスクリーニングの有効性と費用対効果比については,まだ明らかになっていない[Howell et al 2017].
回避すべき薬剤や環境
脛骨異形成やdystrophic typeの側彎症を有する患者に関しては,整形外科医の勧めによって活動が制限されることがある.
NF1患者に対する放射線治療は,照射部位における悪性末梢神経鞘腫瘍の高い発症リスクと関連しているとされている[Evans et al 2002, Sharif et al 2006,Tsang et al 2017].
リスクのある血縁者の検査
リスクのある血縁者の検査については「遺伝カウンセリング」の項を参照のこと.
妊娠中の管理
女性NF1患者の多くは正常な妊娠経過をたどるものの,早産,帝王切開による分娩,妊娠高血圧症,常位胎盤早期剥離の頻度は一般女性に比べて高い[Chetty et al 2011, Terry et al 2013,Leppävirta et al 2018].多くのNF1女性患者は妊娠中に神経線維腫の数や大きさが急速に増大したと訴える.しかし,13人のNF1合併妊婦と同年齢の13人の妊娠していないNF1女性患者について妊娠期間をまたいだ平均4.7年観察したところ,全身MRIにおける皮膚神経線維腫あるいは叢状神経線維腫の体積に継続するような変化はみられなかった[Well et al 2020].
研究段階の治療
種々の重要な細胞内シグナル伝達経路を阻害することでNF1関連のMPNST治療につなげる研究が,モデルを用いた前臨床研究や初期の臨床試験として進行中である[Brosseau et al 2020,Foiadelli et al 2020].
NF1関連の神経膠腫に関する前臨床研究ならびに臨床研究も数多く進行中である[Packer et al 2020,Packer & Vezina 2020].
疾患の根本的な原因であるNF1遺伝子のバリアントを編集しようとする遺伝子治療についても,モデル系での研究がおこなわれている[Cui et al 2020,Leier et al 2020].
NF1に関連して生じる白血病、皮膚神経線維腫、疼痛、便秘、高血圧、認知障害、学習障害,行動障害、社会性の障害、運動障害に関する治療を評価する臨床研究も進行中である.
NF1の臨床試験に関する最新のリストは,米国においてはClinicalTrials.gov,ヨーロッパにおいてはEU Clinical Trials Registerを参照のこと.
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
神経線維腫症1型(NF1)は常染色体顕性遺伝の形式をとる.
家族構成員のリスク
発端者の両親
- 約50%のNF1患者には罹患した親がいる
- 残り約50%はNF1遺伝子に新生(de novo)の病的バリアントが生じたことに起因する.
- 新生(de novo)の病的バリアントによると思われる発端者の両親(両親はいずれもNF1ではないと思われる)についても,下記評価をおこなうことが推奨されている.
- NF1にみられる皮膚やその他の所見に着目した既往歴の聴取および身体診察.
- さらに虹彩小結節や脈絡膜色素斑,その他NF1の眼科的所見を検索するための眼科検査(細隙灯顕微鏡や,近赤外線反射イメージングまたは光干渉断層画像化法(OCT)を含む).
- 既往歴の聴取や身体診察,眼科検査を実施した結果,NF1患者の両親が共にNF1の臨床診断基準を満たさなければ,発端者において新生(de novo)の病的バリアントが生じたことに起因するNF1の可能性がかなり高い[Legius et al 2021].別の可能性としては,NF1モザイクの片親から,あるいは非常に稀ではあるがヘテロ接合性バリアントを有するものの不完全浸透の片親から病的バリアントを受け継いだことにより発端者がNF1になることも起こりうる.小児NF1患者で病的バリアントが同定されていれば,両親に対して同バリアントに限定した分子遺伝学的検査を実施することによって,モザイクの探索や,片親がヘテロ接合性バリアント保有者(ただし不完全浸透により見かけ上は非罹患者)であるかを確認することができる.
注:両親いずれにもNF1の臨床症候がみられず、かつ白血球由来DNAを用いた標準的な分子遺伝学的検査で発端者と同じ病的バリアントが検出されなかった場合でも、親の体細胞モザイクあるいは生殖細胞系列モザイクの可能性は残る[Lázaro et al 1995,Bottillo et al 2010,Trevisson et al 2014,Yang et al 2020]. - 家族がNF1であることを認識していない,あるいは症状や徴候に気づく前に片親が早逝してしまったといった理由で,家族歴が無いように見えることがある.そのため,NF1の徴候についての臨床診査を両親に対して行わない限り,家族歴が無いとは言い切れない.
注:新生(de novo)の病的バリアントに起因するNF1患者の中には,体細胞モザイクによりNF1の症候が限局的に認められる,あるいは非常に軽微なことがある[Messiaen et al 2011,García-Romero et al 2016,Kluwe et al 2020]. NF1遺伝子の病的バリアントをモザイクで有する片親から,疾患が子に遺伝する確率は50%よりも少なくなる.ただし,子が病的バリアントを受け継いだ場合には,全身の細胞に病的バリアントを有することになるため症状が重くなるとがある.
発端者の同胞
発端者の同胞におけるリスクは両親の臨床的状態によって異なる.
- 片親が罹患している場合,同胞におけるリスクは50%である.NF1遺伝子の病的バリアントを受け継いだ同胞はNF1の症状を呈するが,その症状は発端者よりもかなり重症(あるいは軽症)となることがある.
- 発端者の片親がNF1遺伝子の病的バリアントをモザイクで有している場合,同胞が典型的な(大抵の場合はより重症な)NF1を有するリスクは50%よりも少なく,おそらくその数字は親の生殖細胞におけるNF1遺伝子の病的バリアントの割合によって異なってくる.ただし、親の生殖細胞におけるNF1遺伝子の病的バリアントの割合は,親の血液や他の組織における病的バリアントの割合から推測することはできない.
- 慎重な既往歴聴取や身体診察,眼科的検索をおこなっても,両親のいずれもNF1の臨床診断基準を満たさないのであれば,NF1患者の同胞がNF1である可能性は低いが,生殖細胞系列モザイクの可能性があるため一般集団のよりは高い.明らかに罹患していない両親から2人のNF1患者が生まれた例でNF1遺伝子の病的バリアントの生殖細胞系列モザイクが報告されている[Lázar et al 1995,Bottillo et al 2010,Trevisson et al2014,Yang et al 2020].
発端者の子
- 発端者の子はそれぞれ50%の確率でNF1遺伝子の病的バリアントを受け継ぐ.
- 浸透率はほぼ100%であり,NF1遺伝子の病的バリアントを受け継いだ子はNF1の症状を呈する.しかし,子の症状は罹患した親に比べかなり重度なこともあればかなり軽度となることもある.
他の家族構成員
他の血縁者のリスクは発端者の両親の状況によって異なる.発端者の片親が罹患している場合には,その親の血縁者もリスクを有していることになる.
関連する遺伝カウンセリング上の諸事項
同一家系に新生(de novo)の病的バリアントが複数存在する可能性
Upadhyayaらはひとつの家系において3種類の異なるNF1遺伝子病的バリアントが確認された症例を報告しており[Upadhyaya et al 2003],同一家系内の罹患者が同じバリアントを有するとの推測には注意が必要であるとしている.ほかにも,2つの異なるNF1遺伝子病的バリアントがみられた家系が複数報告されている[Klose et al 1999,Pacot et al 2019].
見かけ上は新生(de novo)の病的バリアントと思われる家系についての留意点
常染色体顕性遺伝(優性遺伝)の疾患において発端者の両親がいずれも発端者と同じ病的バリアントを有していない場合や,臨床所見がない場合には,発端者に同定された病的バリアントはおそらく新生(de novo)と考えられる.しかしながら,生物学的父親や生物学的母親が異なる場合(例:生殖補助医療による)あるいは,明かされていない養子縁組など,非医学的な要因が関係する可能性もある.
家族計画
- 遺伝的リスクの評価や出生前診断/着床前診断の可否についての遺伝カウンセリングは,妊娠前の実施が望ましい.
- 罹患しているもしくはリスクがある若い成人に対して遺伝カウンセリング(子に生じる可能性のあるリスクや子を設ける際の選択肢を含む)が実施されることが望ましい.
出生前診断および着床前診断
分子遺伝学的検査
家系内の罹患者にNF1遺伝子の病的バリアントが同定されていれば,リスクのある妊娠に対する出生前診断や着床前診断が可能となる.
超音波検査
非常に重症のNF1を出生前に超音波で診断した報告があるが[McEwing et al 2006],大多数のNF1について超音波検査は有用性が低い.
特に早期診断よりも中絶を目的として考慮される場合は,医療関係者と家族の間では出生前診断に対する見解の相違が生じやすい.多くの医療機関では最終的には両親の意思を尊重するとしているが,この問題については注意深い検討が求められる.
訳注:日本では一般にNF1に対して出生前診断の適応があるとは考えられておらず,着床前診断も行われていない.
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- hildren's Tumor Foundation
95 Pine Street
16th Floor
New York NY 10005
Phone: 800-323-7938 (toll-free); 212-344-6633
Fax: 212-747-0004
Email: info@ctf.org
www.ctf.org - Medical Home Portal
Neurofibromatosis Type 1 - MedlinePlus
Neurofibromatosis 1 - National Organization for Rare Disorders (NORD)
Neurofibromatosis Type 1 (NF1) - Neurofibromatosis Network
213 South Wheaton Avenue
Wheaton IL 60187
Phone: 800-942-6825
Fax: 630-510-8508
Email: admin@nfnetwork.org
www.nfnetwork.org - Nerve Tumours
UKLondon
United KingdomPhone: 0208 439 1234
Email: info@nervetumours.org.uk
www.nervetumours.org.uk - RASopathiesNet
Email: info@rasopathiesnet.org
www.rasopathiesnet.org - NF Registry
Welcome to the NF Registry
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:神経線維腫症1型:遺伝子とデータベース
| 遺伝子記号 | 遺伝子座 | タンパク質 | 座位特異性 | HGMD p | ClinVar |
|---|---|---|---|---|---|
| NF1 | 17q11.2 | ニューロフィブロミン | NF1データベース | NF1 | NF1 |
データは以下の標準的な参照サイトより収載した:遺伝子はHGNCから,染色体遺伝子座はOMIMから,タンパク質はUniProtから.リンク先のデータベース(Locus Specific, HGMD, ClinVar)の説明はこちらを参照のこと.
表:BOMIMにおける神経線維腫症1型関連情報(OMIMですべてをみる)
| 162200 | 神経線維腫症1型;NF1 |
| 613113 | ニューロフィブロミン1;NF1 |
分子遺伝学的な発症機序
NF1遺伝子がコードするニューロフィブロミンは,ras-GTPaseを活性化することで,細胞増殖の制御や腫瘍抑制因子として作用している[Rad & Tee 2016,Bergoug et al 2020].それとは別にニューロフィブロミンには体細胞分裂への関与や,アデニリルシクラーゼ活性と細胞内サイクリックAMP産生の制御といった機能も有している[Mo et al 2022].
NF1は大きな遺伝子で,これまでに3,000種類を超える病的バリアントが報告されている[Bettegowda et al 2021].繰り返し確認されたバリアントは複数あるものの,いずれも家系解析研究で検出されることはほとんどない.大多数の病的バリアントはこれまでに報告例がなく,約半数は新生(de novo)の病的バリアントである.
NF1遺伝子の生殖細胞系列病的バリアントの大部分は,mRNAのスプライシングを変化させることで、遺伝子産物に重度のトランケーション(短縮化)を引き起こしていると考えられる.
疾患を引き起こすメカニズム
機能喪失型である.
NF1遺伝子に特異的な検査上の注意点
スプライスバリアントは標準的なゲノムDNA(gDNA)シーケンシング単独では検出できないことがある.
表7:NF1遺伝子の病的バリアントの中で注目すべきもの
| 参照配列 | DNAヌクレオチドの変化 | 想定されるタンパク質の変化 | コメント[文献] |
|---|---|---|---|
| c.2970_2972 delAAT | p.Met992del | 「遺伝子型-表現型相関」の項を参照 |
|
| c.3112A>G | p.Arg1038Gly | ||
| c.3445A>G | p.Met1149Val | ||
| c.3446T>C | p.Met1149Thr | ||
| c.3447G>A | p. Met1149Ile | ||
| c.3447G>C | p. Met1149Ile | ||
| c.3447G>T | p. Met1149Ile | ||
| c.3826C>G | p.Arg1276Gly | ||
| c.3826_3827delinsGA | p. Arg1276Glu | ||
| c.3827G>A | p. Arg1276Gln | ||
| c.3827G>C | p. Arg1276Pro | ||
| c.3827G>T | p. Arg1276Leu | ||
| c.4267A>C | p.Lys1423Gln | ||
| c.4267A>G | p. Lys1423Glu | ||
| c.4268A>C | p. Lys1423Thr | ||
| c.4268A>T | p. Lys1423Met | ||
| c.5425C>T | p.Arg1809Cys | ||
| c.5425C>G | p.Arg1809Gly | ||
| c.5425C>A | p.Arg1809Ser | ||
| c.5426G>C | p.Arg1809Pro | ||
| c.5426G>T | p.Arg1809Leu | ||
| NG_009018.1 | 1.4Mbの欠失1 | -- |
表に記載されているバリアントは,著者によって提供されたものである.GeneReviewsのスタッフは,バリアントの分類を独自には検証していない.
GeneReviewsは,Human Genome Variation Society (varnomen.hgvs.org) の標準的な命名規則に従っている.命名法についての説明はQuick Referenceを参照のこと.
- 1型欠失と呼ばれるNF1遺伝子全体の欠失
癌および良性腫瘍
NF1患者において発症頻度の高いいくつかの腫瘍に関しては,NF1の臨床症状を持たない患者がNF1遺伝子の片アリル,あるいは両アリルに体細胞バリアント(生殖細胞系列バリアントではない)を有することがある[D'Angelo et al 2019,Eoli et al 2019,Dunnett-Kane et al 2020,Fisher et al 2021].NF1遺伝子の体細胞病的バリアントによる散発性腫瘍の例としては,悪性末梢神経鞘腫瘍,褐色細胞腫,若年性慢性骨髄性白血病,神経膠腫,乳癌がある.なお,体細胞のNF1遺伝子の病的バリアントは脂肪肉腫,肺腺癌,卵巣癌,大腸癌,膀胱移行上皮癌,神経芽腫,悪性黒色腫,および成人急性骨髄性白血病でも認められるが,これらがNF1患者に生じることは稀である [Philpott et al 2017, Dunnett-Kane et al 2020]. NF1遺伝子の体細胞バリアントによる癌は,NF1遺伝子の生殖細胞系列バリアントを有する患者にみられる癌とはバリアントのスペクトルが異なることが多い。
更新履歴:
- Gene Review著者: J M Friedman, MD, PhD
日本語訳者: 大畑 尚子(沖縄県立中部病院 総合周産期母子医療センター)
Gene Review 最終更新日: 2009.6.2. 日本語訳最終更新日: : 2011.10.17 - Gene Review著者: J M Friedman, MD, PhD
日本語訳者: 江田肖(瀬戸病院 遺伝診療科),大畑尚子(沖縄県立中部病院 総合周産期母子医療センター)
Gene Review 最終更新日: 2012.5.3 日本語訳最終更新日: 2014.3.18 -
Gene Review著者: J M Friedman, MD, PhD
日本語訳者: 森川真紀(名古屋大学医学部附属病院ゲノム医療センター),生田国大(名古屋大学医学部附属病院ゲノム医療センター/整形外科)
Gene Review 最終更新日: 2019.6.6 日本語訳最終更新日: 2021.1.12 -
Gene Reviews著者: J M Friedman, MD, PhD
日本語訳者: 佐藤康守(たい矯正歯科),森川真紀(名古屋大学医学部附属病院ゲノム医療センター),生田国大(名古屋大学医学部附属病院整形外科)
GeneReviews最終更新日: 2022.4.21. 日本語訳最終更新日: 2023.11.20.[in present]
![]()