神経線維腫2型
(Neurofibromatosis 2)
[同義語:Bilateral Acoustic Neurofibromatosis, Central Neurofibromatosis, NF2, Neurofibromatosis Type II]
Gene Reviews著者: D Gareth Evans、 MD、 FRCP
日本語訳者: 小川千穂(ボランティア翻訳者),櫻井晃洋(信州大学医学部附属病院遺伝子診療部)
Gene Reviews 2011.8.18. 日本語訳最終更新日: 2012.1.2
要約
疾患の特徴
者は30歳までに両側性前庭神経鞘腫を発症する。罹患者はほかの脳神経や末梢神経の神経鞘腫、髄膜腫、上衣腫やごくまれにではあるが、星状細胞腫を発症することもある。視覚的に重大な白内障に進行することはほとんどない後嚢下白内障が最もよく見られる眼科的病変であり、NF2の最初の兆候である可能性がある。小児期に発症する単神経炎は次第に認知されつつある。これは持続的な顔面麻痺、斜視(第Ⅲ神経麻痺)、もしくは垂手や下垂足がしばしば認められる。
診断・検査
NF2の診断は臨床的基準に基づいて行われる。 NF2は本症に関連していることが知られている唯一の遺伝子である。シークエンス解析もしくは変異スキャンと欠失/ 重複解析の組み合わせを含むNF2分子遺伝子検査は、家族歴陽性ではあるが家系内の発端者が明らかではないほとんどの罹患者の変異を検出する。
臨床的マネジメント
病変に対する治療 :
前庭神経鞘腫の治療の第一選択は手術である。定位的放射線治療というガンマナイフを用いられることがもっとも一般的である療法が、手術の代わりになりうる。前庭神経鞘腫患者は平衡感覚や水中での方向感覚の喪失という問題が知らぬ間に進行することを意識する必要がある。溺死する可能性があるからである。聴覚喪失に対する治療には聴覚訓練士への紹介、読唇術や手話の指導、補聴器やもしくは人工内耳や脳幹インプラントがある。
経過観察
:罹患者もしくはリスクのあるものが対象
毎年のMRI検査を10歳から12歳ぐらいに開始し少なくとも30歳代まで続ける。脳幹聴覚誘発反応テストを含む、聴覚評価は有用である。
回避すべき薬剤や環境 :
NF2関連腫瘍の放射線治療は、特に小児の場合、腫瘍の進行を促したり、形質転換させることがある。
リスクのある親族への検査 :
家族性NF2変異を遺伝している親族を早い段階で発見することにより、病気の徴候を早期発見・治療をすることができ、適切な経過観察をすることが可能になる。
遺伝カウンセリング
NF2は常染色体優先遺伝の形式をとる。患者の約50%が罹患した親からNF2を受け継ぐが、残りの50%は新生突然変異が原因である。しかし、散発例(家族内でひとりだけが罹患)の25%から30%はNF2遺伝子変異をモザイクで有する。発端者の家族に他の罹患者がいれば、発端者の子供は50%の可能性で変異を受け継ぐ。リスクのある妊娠に対する出生前診断は、家系内での遺伝子変異や連鎖が明らかとなっていれば可能である。
診断
臨床診断
NIHのNF2に対する診断基準の改定版は、発端者(家系内で罹患が明らかな最上の世代の患者)のより早期診断を可能としている。 NF2の臨床診断基準改定では特異度に影響を及ぼすことなく検出感度を向上させた。改定された臨床基準によると、以下の所見に1つでもあてはまるとNF2と診断される。
- 両側性の前庭神経鞘腫
- 第一度親近者にNF2罹患者がおり次のいずれかが認められる
片側性前庭神経鞘腫 または
以下のうち2つ*:髄膜腫、神経鞘腫、神経膠腫、神経線維腫、後嚢下白内障
- 片側性前庭神経鞘腫と以下のうちの2つが認められる*
髄膜腫、神経鞘腫、神経膠腫、神経線維腫、後嚢下白内障
- 多発性髄膜腫と次のいずれかが認められる:
- 片側性前庭神経鞘腫
- 以下のうちの2つ*:神経鞘腫、神経膠腫、神経線維腫、白内障
*以下のうちの2つ*:2種の腫瘍もしくは白内障
検査
染色体分析
種々の染色体異常がNF2と関連することがある。しかし通常の細胞遺伝学的解析で検出できるような大きな染色体の変化はきわめてまれである。
- NF2を含む細胞遺伝学上明らかな欠失は知的障害や先天性異常をおこすことがある。
- 22番環状染色体はNF2の診断基準を満たす多発性髄膜腫や前庭神経鞘腫をおこすことがある。 NF2遺伝子座自体は通常、環状染色体内に存在するが、環状自体が不安定なためしばしば消滅する。
- NF2を分断するあきらかな均衡型染色体転座がNF2の原因であるとも報告されている。
FISH法
FISH法によりNF2や遺伝子全体から複数のエクソンが失われる小規模な欠失を特定できる。
分子遺伝学的検査
GeneReviewsは,分子遺伝学的検査について,その検査が米国CLIAの承認を受けた研究機関もしくは米国以外の臨床研究機関によってGeneTests Laboratory Directoryに掲載されている場合に限り,臨床的に実施可能であるとする. GeneTestsは研究機関から提出された情報を検証しないし,研究機関の承認状態もしくは実施結果を保証しない.情報を検証するためには,医師は直接それぞれの研究機関と連絡をとらなければならない.―編集者注. .
遺伝子 NF2はその変異株がNF2の原因と明らかになっている唯一の遺伝子である。
臨床検査
表1.NF2で用いられる分子遺伝学的検査の概要
| 遺伝子 | 検査法 | 検出される変異 | 変異検出率1 | 検査の利用可能性 |
|---|---|---|---|---|
| NF2 | シークエンス解析/変異スキャン2 | シークエンスバリアント3 | 脚注4と5を参照 | 臨床 |
| 欠失/ 重複解析6 | 部分的かつ全体的な遺伝的欠失7 | 脚注8を参照 | ||
| 連鎖解析9 | なし | なし |
検査の利用可能性はGeneTests Laboratory Directoryを参照。Gene ReviewではUS CLIA-licensed laboratoryもしくはnon-US clinical laboratoryによってGene Tests Laboratory Directoryに記載されている場合のみ、分子遺伝学的検査は臨床的に利用可能と位置付けている。GeneTestsは研究機関から提出された情報を検証しないし、研究機関のライセンスもしくは実施結果を保証していない。情報を検証するためには、医師はそれぞれの研究機関に直接問い合わせること。
- 散発例とNF2保因家族の第一世代の発端者においては体細胞モザイクである可能性が高いため、変異検出率はより低かった。(分子遺伝学的検査のテスト結果の解説を参照)
- 変異スキャンを用いたNF2変異検出率は全体の2/3にまで達し、シークエンス解析に匹敵する。
- シークエンス解析による変異検出例としては、微細な遺伝子内欠失や挿入、ミスセンスやナンセンスやスプライス部位の変異を含む。
- 変異スキャンは単一エクソンの欠失/重複解析を組み合わせると、変異検出率は散発例で72%に向上し、家族例では92%をこえる。
- 他の報告ではより低い変異検出率が示されているが、これは体細胞モザイクを伴うより軽症な例も含んでいるためであろう。約25-33%の変異は体細胞モザイクのために検出されない。モザイクレベルで10%以上の変異はリンパ球DNAにより検出されることがある。残りのモザイク変異の同定には腫瘍組織を用いた検査が、通常必要になる(分子遺伝学的検査のテスト結果の解説を参照)。
- ゲノムDNAのコーディング領域や隣接イントロン領域をシークエンス解析により迅速には検出できない欠失/重複解析や定量PCR、ロングレンジPCE、MLPA法、アレイCGH(遺伝子/セグメント特異的)を含むさまざまな方法が用いられることがある。
- 全ゲノムの欠失・重複解析するアレイCGHは、この遺伝子/セグメントを含むことがある。詳細はアレイCGHの項を参照のこと。
- 多くの欠失、または頻度は低いが、単一もしくは重複エクソンはMLPA法により検出できる。
- NF2生殖細胞変異家系の少なくとも10-15%に10-600kbの欠失が認められる。 しかし、遺伝ケースにおいては、約20%に認められ、シークエンス解析と欠失・重複解析との組み合わせると93%まで感度をあげることができる(101/108)。
- NF2変異がなくても、少なくとも異なる世代で2人の罹患者がいる家系においては連鎖解析が適応されることもある。 連鎖解析は罹患した家族の正確なNF2臨床診断と家族における正確な遺伝的関係の把握にもとづいて行われている。それゆえ連鎖解析は検査をうける家族の協力と意思が大事になる。
NF2連鎖解析に用いられるマーカーはNF2遺伝子との非常に強い関連性がある。したがって、95%以上の家系で、99%以上の精度で行うことができる。連鎖解析は患者に新生突然変異が起こり、家系に他の罹患者がいない場合など、通常、患者が1人だけの家族には行えない。しかし、生殖細胞および腫瘍組織DNAを用いた修正連鎖解析では腫瘍組織で失われたアレルを引き継がない散発例の子孫においてはNF2を排除することができる。
検査結果の解釈
- シークエンス解析の結果解説の考察点についてはこちらを参照のこと。
- 研究によれば、新生突然変異によるNF2罹患者の最大25-33%は体細胞モザイクである。NF2病原性変異の体細胞モザイクを有する罹患者の同定は以下の理由で困難である。
- 両側性前庭神経鞘腫を発症していないことがある。
- リンパ球のようにNF2病変を伴わない分子遺伝検査では異常がみつからないことある。よって腫瘍組織の分子遺伝検査が体細胞モザイクの存在を証明するためには必要である。
- もし子がモザイクであることが確認されれば、両親がNF2である可能性は否定できるが、逆に子に変異がなかったとしても、両親がモザイクの可能性を否定する根拠にはならない。
特定のアレル多型についての情報は分子遺伝学情報である以下を参照 Table A。
検査手順
発端者の確定診断をするために以下2つのサンプル型のうちの片方が用いられる。
- 罹患者の親をもつ者の白血球DNA
- 散発例で発病した罹患者の腫瘍組織DNA
生殖細胞変異を検知する分子遺伝学的検査は下記の手順で行われる
- MLPA法のような技術を用いた大規模な欠失の検査
- エクソン1-15のシークエンス解析(エクソン16-17の変異はこれまでに報告されていない)
腫瘍組織DNAが検査されれば、NF2の両アレルの変異は同定されるはずである。
- このことはヘテロ接合性の喪失(LOH)を評価することにより一方のNF2アレルの欠失(もしくは不活化)を検査することを意味する。
- 両方のNF2変異アレルが腫瘍内で確認されれば、白血球DNAで生殖細胞変異であるか体細胞変異であるか確定できる(腫瘍内に存在するときのみ)。
予測検査
- 家系内にNF2罹患者(例:発端者)が確認されているものの、遺伝状況が明らかでないリスクある親族はNF2変異(生殖細胞変異か体細胞変異であるか)の有無を検査できる。
- NF2変異が確認されない稀なケースの場合、異なる世代に少なくとも2人の罹患者のいる家族では連鎖解析が用いられるか、もしくは散発例の子孫の遺伝状態を明らかにするために腫瘍組織DNAが用いられることがある。
リスクのある妊娠に対する出生前診断や着床前診断を実施するにあたっては、事前にその家系で原因となる遺伝子変異を同定しておく必要がある。
注:GeneTestsLaboratoryDirectoryに記載されている施設で実施可能な臨床用途を含むことがGeneReviewの方針である。しかし必ずしも著者、編集者、評価者による推奨を反映するものるものではない。
遺伝的に関連する(アレルと関連した)疾患
NF2変異と関連していると知られている他の表現型はない。
臨床像
NF2患者で症状が現れる平均年齢は18-24歳である(発症年齢は0歳から70歳に分布する)。患者のほぼ全員が30歳までに両側前庭神経鞘腫を発症する。前庭神経鞘腫に加え、NF2患者は他の脳神経や末梢神経の神経鞘腫、髄膜腫、上衣腫、さらにまれではあるが星状細胞腫を生じることもある。
腫瘍の大きさ、部位、数などにより、NF2患者の臨床像は個人差が大きい。これらの腫瘍は悪性ではないが、腫瘍が発生する解剖学的位置や多発性のために病状は重く、早期死亡の原因にもなる。平均死亡時年齢は36歳である。正確な診断から死亡までの平均生存期間は15年である。早期診断と専門医療施設でのよりよい医療が生存率を向上させる。
NF2は成人発症型疾患とみなされているため、皮膚腫瘍や眼球所見が初発症状となりうる小児では、十分に認識されずに見過ごされている可能性がある。Evansらによる英国のNF2患者120名の臨床症状を表2に示す(この研究では皮膚腫瘍や白内障は初発症状に含めていない)。
表2.NF2患者120名の臨床症状
| 症状 | 患者の比率 |
|---|---|
| 片側性難聴 | 35% |
| 局所性筋力低下 1 | 12% |
| 耳鳴 | 10% |
| 両側性難聴 | 9% |
| 平衡障害 | 8% |
| けいれん | 8% |
| 局所性感覚障害 | 6% |
| 失明 | 1% |
| 無症状、家族スクリーニングで発見 | 11% |
- 脊髄腫瘍、単神経障害、多発神経障害が原因となる。
前庭神経鞘腫
初発症状には耳鳴、難聴、平衡障害がある。症状は通常出現時期がはっきりしないが、時には突然の難聴が起きることもあり、これはおそらく腫瘍による血行障害によるものと考えられる。患者はしばしば一方の耳で電話が聞き取りにくいことや、夜間あるいは平坦でない道を歩くときの不安定さを訴える。時間が経過すると腫瘍は前庭から内側の小脳橋角部に進展し、治療がなされなければ脳幹圧迫と水頭症をきたす。腫瘍が大きい場合も重篤な顔面麻痺をきたすことはまれである。神経鞘腫は他の脳神経や末梢神経に発生することもあり、運動神経より感覚神経によりできやすい。
脊髄腫瘍
少なくとも2/3のNF2患者で脊髄腫瘍が発生し、しばしば本症において最も深刻で治療が困難な病変となる。 最もよくみられる脊髄腫瘍は神経鞘腫であり、通常脊椎管内で後根に発生し、内側と外側の両方向に進展する結果、ダンベル型の形態をとる。星状細胞腫や上衣腫のような脊髄の髄内腫瘍はNF2患者の5-33%に発生する。脊髄障害を発症した患者のほとんどは多発性腫瘍である。多発性腫瘍はしばしば画像診断で見つかるが、NF2患者の多くはそれによる症状を現さない。
髄膜腫
横断的研究からNF2患者の約半数に髄膜腫が生じることがわかった。 しかし、一生涯では80%に達することがわかってきた。多くは頭蓋内であるが、脊髄で生じることもある。NF2髄膜腫は頭蓋底よりもテント上にできることが多く、通常、線維芽細胞様種である。眼窩内髄膜腫は視神経圧迫の結果失明の原因となることがある。また頭蓋底に生じた場合は脳神経麻痺や脳幹圧迫、水頭症の原因となる。髄膜腫は特に小児期にNF2の特徴として現れることがある。
Genotype-Phenotype Correlationsを参照
眼病変
NF2患者の約1/3では片眼あるいは両眼の視力低下をきたす。重篤な白内障にはめったに移行しない後嚢白内障の白濁がもっともよく見られる眼病変である。
水晶体の白濁は前庭神経鞘腫による症状が現れる以前から出現し、小児でも認められる。出生時に白内障の症状があり、発育時にはこれらの小児では弱視はよく見られる症状である。
網膜過誤腫や網膜上膜は1/3以下の患者に認められる。まれに他の眼病変を生じることもある。第一次硝子体過形成遺残症(PHPV)の父子例が報告されている。 成人期に角膜に特別な問題がおきると、殊に術後に顔面神経、三叉神経や中間神経機能を消失する。頭蓋内腫瘍や眼窩内腫瘍は視力低下や複視の原因となる。
単/多発神経障害
特に小児のNF2患者に認識される特徴には単神経障害があるが、これはしばしば部分的にしか回復しない顔面麻痺、斜視(第III神経麻痺)や垂手、下垂足といった症状を呈する。 下垂足はポリオに似た症状を呈する。
直接腫瘍と関係しない成人の進行性多発神経障害も認識が高まっている。
小児の単神経障害と成人の多発神経障害に関するさらなるエビデンスが腓腹神経生検から得られている。
その他
NF1で見られるのと類似した腎血管病変が1例で報告されている。
NF2遺伝子変異の体細胞モザイク
モザイクの可能性が疑われるのは片側性前庭神経鞘腫や他の多発性腫瘍であるが、頻度が高いのは同側性の腫瘍がある患者である。
これは、多発性腫瘍で解析されたDNAにおいてほとんどの例で確認されている。病理
NF2の腫瘍はシュワン細胞、髄膜細胞、グリア細胞に由来する。これはいずれも良性である。NF2の前庭腫瘍の約40%は小葉構造をとるが、これはNF2の散発例では通常みられない。
- NF2関連前庭神経鞘腫は非NF2腫瘍に比べてより浸潤性であり、高分化を認める。
- NF2関連髄膜腫は非NF2髄膜腫より高分化である。NF2患者に生じた髄膜腫は通常線維芽細胞様種である。
- 神経膠腫におけるNF2を持つ患者と持たない患者では組織学的に違いはない。
遺伝子型と臨床型の関連
家族内での変異差は家族間のものよりも小さいということは、根本的な遺伝子型が罹患者の臨床型に大きな影響を与えていることを示している。
NF1と異なり、NF2遺伝子の大規模欠失例では臨床像はより軽症である。 NF2生殖細胞変異家系の少なくとも10-15%に10-600kbの欠失が認められる。これらはたとえ大きな欠失の場合でも精神発達遅滞は伴わない。
NF2生殖細胞変異のタイプは、 NF2関連頭蓋内髄膜腫、脊髄腫瘍、末梢神経腫瘍の発生数にかかわる重要な決定因子である。
- 遺伝子内の位置に関係なく、ナンセンス変異やフレームシフト変異では臨床像はより重症となる。
- スプライス部位の変異は軽症型、重症型のいずれとも関連しており、 3’側に変異がある時はより軽症である。
- ミスセンス変異は通常軽症で、NF2ではしばしば最も軽症である。
- トランケーション変異は早期に発病し多くのNF2関連の頭蓋内髄膜腫、脊髄腫瘍、末梢神経腫瘍に関連している。一般に、トランケーション変異(フレームシフトやナンセンス)はミスセンスやスプライス部位の変異、もしくは欠失よりも、疾患に関連した死亡率も高い。トランケーション変異は脊髄腫瘍の有病率も高くなる。これらの変異により産生されるタンパクの大部分はナンセンス変異依存分解系により分解されて失われると考えられるので、これらの変異に影響を与える明らかな優性阻害についてはさらなる調査が必要である。
- NF2の5’側にある変異よりも3‘側にある変異(特にエクソン14-16)の方が髄膜腫罹患に対してリスクが低い(図1)。
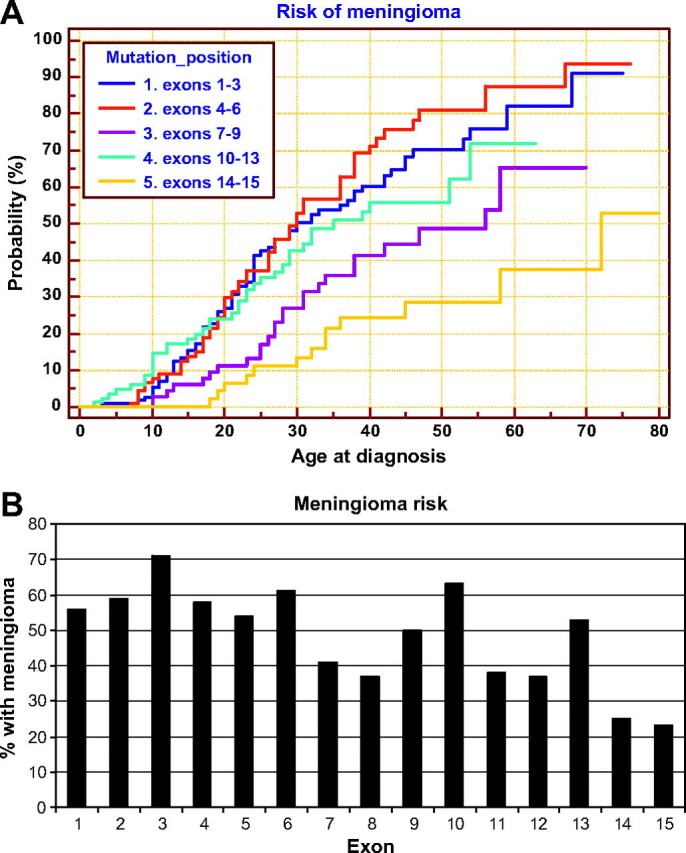
図1 NF2遺伝子の変異の位置が髄膜腫発生頻度に影響を与える
A.各機能ドメイン別に髄膜腫のリスクを示したKaplan-Meier曲線。NF2遺伝子をエクソン1-3、4-6、7-9、10-13、14-16の領域に分けてある。
B.個々のエクソンに変異がある場合の髄膜腫発症リスク。5‘側は3’側よりもリスクが大きく、機能ドメインの境界領域の変異はその他の領域に比べてリスクが高い。
- 重症のNF2を引き起こすような典型的なトランケーション変異の体細胞モザイクでは(リンパ球DNAで検出されたときでさえ)臨床像は軽度であることがある。
浸透率
NF2の浸透率は100%である。病原性生殖細胞変異を有している者は全員が、事実上、生涯の間に発症する。
発症年齢は変異のタイプによって異なる。 Genotype-Phenotype Correlationsを参照。
促進現象
NF2の促進現象を示唆する報告もあるが、おそらく第一世代にNF2変異モザイクを有した軽症例があり、第二世代以降で生殖細胞変異を有した結果、重症型の臨床像を呈したものである。
病名
「神経線維腫症(neurofibromatosis)」という病名は、本症の主要病変が神経鞘腫と髄膜腫であることを考えれば誤った命名である。前庭神経鞘腫(以前は聴神経腫と呼ばれていた)は、かつてはフォン・レックリンハウゼン病、つまりNF1の部分症と考えられており、このためにNF2罹患者における多くの事例がNF1罹患者の中に含められていた。1987年からは、大部分の報告はNF1とNF2を正確に区別してきており、NF2は両側性聴神経線維腫症もしくは中枢神経線維腫症と説明されている。
頻度
NF2の罹病率は当初33,000-40,000人あたり1人と報告されていたが、実際の頻度はそれより少々低く200,000人あたり1人程度である。
2010年の研究に基づくと、当初予想していた60,000人に1人の罹患率よりは高いが、33,000人に1人ということが確定しつつある。
注:2005年、最近の報告では誤って最大では25,000人あたり1人程度と示された。
NF2の発症頻度に人種差はない。
鑑別診断
本稿で扱われる疾患に対する遺伝学的検査の実施可能性に関する最新情報は,GeneTests Laboratory Directoryを参照のこと.―編集者注.
神経線維腫症1型(NF1)
NF1とNF2は臨床的に別の疾患であり、異なる染色体上の異なる遺伝子の変異による疾患であるが、診断上の混乱が今も続いている。したがって、NF1とNF2を区別するいくつかの所見を記載しておくことが有用であろう。
- NF2患者はNF1患者にみられるような認知的問題(精神遅滞や学習障害)はみられないし、Lisch結節(虹彩過誤腫)も認めない。
- NF2患者の神経鞘腫は、めったにないが、もし万が一あれば悪性化して神経線維肉腫となる。
- よくある誤解とは異なり、NF2患者は一般集団よりは多いかもしれないが、カフェ・オレ斑の数は少ない。
- ダンベル型の神経根腫瘍は、NF2では神経鞘腫であり、NF1においては神経線維腫にあたるものであるが、しばしば最初の診断時において両者の混乱を引き起こす。
神経鞘腫症(Schwannomatosis)はNF2診断の特徴である前庭腫瘍を伴わない多発性神経鞘腫である。以前はこの病名には多発性神経線維鞘腫や多発性神経鞘腫、神経線維鞘腫症などともよばれていた。
神経鞘腫症患者では頭蓋内や脊髄神経根、末梢神経に腫瘍を生じる。悪性化はめったにない。1/3の神経鞘腫症患者においては、腫瘍は解剖学的に限局した位置に発生し体節性の疾患であることを示唆している。
家族例では常染色体優性遺伝を示し、病態は多様で浸透率は不完全である。 多発神経鞘腫の一部の患者はNF2の診断基準を満たし、神経鞘腫の散発例の一部にもNF2変異のモザイクを認めるものの、神経鞘腫症は臨床的にも遺伝学的にもNF1やNF2とは別のものである。SMARCB1(1NI1)の変異が神経鞘腫症患者家族で確認された。その後の分析によって、家族性神経鞘腫症の30-60%はSMARCB1の変異によって発症していることが示されたが、しかしごくわずかな散発例にすぎない。
片側性前庭神経鞘腫は一般集団においてもよくみられる腫瘍で、全頭蓋内腫瘍の5-10%を占め、大部分の小脳・橋角部腫瘍を占める。前庭神経鞘腫のうち両側性でNF2に関連しているものは5%にすぎず、残りの95%は片側性で遺伝学的にNF2と関係ない患者に生じる。片側性腫瘍がNF2の初発症状である可能性は患者の年齢に依存する。
- 片側性前庭神経鞘腫の臨床症状を伴う30歳未満の患者は、反対側にも腫瘍やNF2を生じる可能性が高いので、綿密にモニターする必要がある。実際明らかに散発性と思われる前庭神経鞘腫患者の6%はNF2遺伝子変異のモザイクである。
- 片側性前庭神経鞘腫を30歳以上で発症した患者はNF2を発症する可能性はかなり低い。
散発性、片側性の前庭神経鞘腫を生じた患者の子がNF2や片側性前庭腫瘍のリスクが高いことはない。単発性の前庭神経鞘腫ではNF2遺伝子の体細胞変異がほぼ常に生じている。; しかしながら、22番染色体の他の遺伝子変異が神経鞘腫の発生に関与している可能性もある。
髄膜腫
典型的な多発性髄膜腫は高齢者に発症するが、25歳未満での単発性髄膜腫では遺伝学的原因の評価を行うべきである。 NF2では前庭神経鞘腫発症以前に髄膜腫を生じることもあるので、小児に発生した髄膜腫ではNF2の初期症状である可能性を考慮する必要がある。多発性髄膜腫はしばしば前庭神経鞘腫を伴わないNF2変異のモザイクに起因している。しかし一般的に前庭神経鞘腫を伴わない成人の多発性髄膜腫がNF2によるものである可能性は低い。常染色体優性遺伝とは区別した前庭神経鞘腫を伴わない多発性髄膜腫のまれな症例が報告されている。最近、神経鞘腫症をある割合で発症させるSMARCB1の変異が一家族で報告されたが、多発性髄膜腫の患者の大部分はSMARCB1変異をもっていない。一家系での連鎖解析では遺伝子座はNF2遺伝子座とは別のものが関係していた。また、散発性髄膜腫の約60%以上ではNF2以外の遺伝子の関与が示唆された。
臨床医への注釈:この疾患に関連した患者特有の同時診療(simultaneous consult)についてはSimulConsult(R)が有用である。患者の症状に基づいた鑑別診断を行う双方向性の診断支援ソフト(登録か組織のアクセスが必要)である。臨床的マネジメント
最初の診断時における評価
NF2との診断を受けた患者では、疾患の程度を明らかにするため、以下の検査が勧められる。
- 頭部MRI
- 脳幹聴覚誘発反応試験を含む聴力検査
- 眼科診察
- 皮膚科診察
- 遺伝カウンセリング
注:NF2患者の評価、治療は本症の多様な合併症に対して臨床的マネジメントについて豊富な経験を持つ専門機関で行われるのが最良である。
- アメリカのNF専門家はこちらを参照www.ctf.org.
- イギリスにおいては4つの専門機関が指定されており、こちらを参照www.specialisedservices.nhs.uk.
症状に対する治療
前庭神経鞘腫
未治療の腫瘍は増殖がゆっくりで短期的には積極的な治療を要しない。治療の原則は手術である。
- 小管間に完全に限局している1.5 mm未満の小さい前庭腫瘍では、聴覚や顔面機能を保存したままの完全摘出も可能である。
- より大きい腫瘍では腫瘍による脳幹圧迫、難聴の悪化あるいは顔面神経障害を生じた時に待機的な減量手術・減圧手術を行うのが恐らく最良である。しかし患者の聴覚に問題がないときは顔面機能を保存したまま、早期に手術を行うか、もしくは手術のタイミングを遅らせるかの選択は難しい。
定位的放射線治療というガンマナイフを用いられることがもっとも一般的である療法が、一部の前庭神経鞘腫に対する手術の代替治療として提供されている。しかしながらNF2患者に対する治療成績は散発性片側性前庭神経鞘腫のそれほどよくはなく、長期的に腫瘍を制御できるのは60%程度である。
頻度は高くないが、ときに悪性化をきたす。しかし放射線照射後の腫瘍増殖には15年を要することもあることを認識しておく必要がある。このことは放射線照射後の部位において、治療病変で再発、もしくは新しい悪性腫瘍(例:膠芽細胞腫)が発症する可能性を意味している。
前庭神経鞘腫患者のマネジメントでは平衡感覚や水中での方向感覚の喪失という問題が知らぬ間に進行することを意識する必要がある。溺死する可能性があるからである。
他の腫瘍
その他の頭蓋内、脳神経、脊髄神経腫瘍は増殖がゆっくりであり、症状を呈していない腫瘍に対する外科手術は障害の出現をかえって早めてしまう可能性がある。
非NF2患者における上衣腫の最適な治療方法としては完全に切除することであり、時に放射線療法や化学療法も行われるが、NF2患者の上衣腫に対しては積極的な治療が有益であるかどうかは明らかでない。
腫瘍抑制遺伝子の機能喪失を伴う患者(特に小児)に対する放射線照射は腫瘍を誘発、進行を加速させたり悪性化させたりする可能性があるので、NF2関連腫瘍に対する放射線療法には注意深い検討が必要である。
聴力
NF2のマネジメントでは聴覚の維持と増強が重要である。すべての患者と家族は最適な聴力と発生訓練を受けるために専門の聴覚訓練士に紹介されるべきである。
- 読唇術は訓練によって上達できる。
- 手話は患者が聴覚を失う前の方がより効果的に習得できる。
- 病気の初期のうちに補聴器を導入するのは有用かもしれない。
- 聴覚を失った患者に対しては蝸牛もしくは脳幹への人工内耳埋め込みによる聴覚リハビリテーションについて検討すべきである。まれではあるが、蝸牛への血行障害をきたしているがその他に神経損傷がない患者では、蝸牛埋め込み術が有用であろう。
眼病変 NF2の他の臨床病変による視覚障害を早期に認識し、マネジメントを行うことは非常に重要である。
二次病変の予防
治療は二次病変の予防に集中する。専門家による適切な腫瘍治療によって、病変によるかなりの後遺症は予防される。
- 麻酔下での処置による合併症を予防するために、頭部手術の前に頸髄のスキャンを行うべきである。
- 脊髄腫瘍は硬膜外麻酔を困難にするので、局所麻酔を行う前に腰仙部の画像検査を行うべきである。
経過観察
家族内でNF2変異が明らかな場合や分子遺伝学的検査で遺伝状態が明らかにできなかった高リスクの人に対しては、以下を行う。
- MRI検査は通常10-12歳で開始するが、発症がもっと遅いことがわかっている家系ではより遅くすることもできる。MRI検査は少なくとも30歳代まで毎年続ける。より早期開始すること(つまり10代より前に頭部MRIを行うこと)の意義は不明であり、いつになれば安全に終了できるかも明らかではない。 50歳代まで無症状であるNF2患者も中にはいるが、若年時に施行するMRIが無症候性腫瘍を発見できる可能性がある。
- 脳幹聴覚誘発反応試験を含む聴力検査はMRIによる画像診断が可能になる前は聴覚神経機能の変化を検出するうえで有用である。
所定の精密な眼科検査は経過観察の一環としてすべてのNF2患者に対して行われるべきである。
回避すべき薬物や環境
NF2の小児に対する放射線照射は避けるべきである。
リスクのある親族に対する検査
リスクある若年の家族に対してNF2分子遺伝学的検査を検討することは適切である。(Genetic Counselingを参照)
- 家系内のNF2変異保有者を早期に同定することは、適切なMRI画像検査や聴覚評価のための聴覚脳幹聴覚誘発反応検査を可能にし、疾患の早期発見と予後の改善につながる。
- NF2変異を受け継いでいない家族を早期に同定することによってMRIや聴覚脳幹聴覚誘発反応検査といった費用のかかる検査を行う必要性がなくなる。
リスクのある親族の検査に関連する遺伝カウンセリングの問題については、遺伝カウンセリングの項を参照のこと。
妊娠管理
妊娠中に神経鞘腫のサイズが増大するという説得力のあるエビデンスはないが、髄膜腫に対してホルモンが影響する可能性はある。そのため、頭蓋内圧が増大するというリスクを評価することが妊娠を考えている女性に対しては重要になる。
研究中の治療法
NF2関連腫瘍に対する効果的な治療法の探求が続けられている。
第一世代の薬の一つとしてはPAK-1阻害薬があげられる。 細胞外シグナル制御キナーゼ1(ERK1)、プロテインキナーゼB(AKT)、インテグリン/焦点接着キナーゼ(FAK)/Src/Rasシグナル伝達経路、血小板由来増殖因子β受容体、ホスファチジルイノシトール3キナーゼ/プロテインキナーゼC/Src/c-Raf伝達回路、血管内皮増殖因子(VEG-F)などを標的とするものや、ほかの経路のもの、そしてアバスチン、エルロチニブ、ラパチニブ、ソラフェニブなどのような薬がNF2の治療に効果的であろう。これらの薬はNF2モデルマウスを対象に動物実験がなされ、北米とイギリスでは最初の臨床試験が始まっている。
もっとも有望な結果はアバスチンによる短・中期間治療であり、これは髄膜腫には効果はないものの急速に増大する神経鞘腫や場合によっては上衣腫にも効果的であるようである。治療薬のさらなる開発は継続中である。
最近の臨床試験ではベバシズマブが有望な治療薬とみなされている。
種々の疾患に対する臨床試験に関する情報はClinicalTrials.govを参照のこと。
その他
遺伝の専門科が配置された遺伝クリニックは患者や家族に自然経過や治療、遺伝形式、他の家族への遺伝リスクだけでなく、利用可能な民間の情報資源に関する情報を提供できる。GeneTests Clinic Directoryを参照。
本疾患に対する疾患特異的または包括的支援組織に関する民間の情報資源を参照。これらの組織は患者と家族に情報、支援、他の患者への連絡情報などを提供するために設立されてきた。遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
NF2は常染色体優性遺伝形式をとる。
患者家族のリスク
発端者の両親
- 約50%のNF2患者には罹患した親があり、残りの50%は新生突然変異の結果として変異遺伝子を有している。しかしながら、25-33%の散発例(つまり、NF2の家族歴がない患者)はNF2遺伝子の変異をモザイクで有している。
- 明らかに新生突然変異による発端者の両親に対する検査として推奨されるものは、既往歴の聴取を行い、もしNF2を疑わせる要素があるならばMRI検査を行う。もし子供がモザイクであることが確認されれば、両親がNF2である可能性は否定できるが、逆に子供に変異がなかったとしても、両親がモザイクの可能性を否定する根拠にはならない。発症年齢は家系内で一定しているので、無症状な両親に対して経過観察を行う必要はない。
発端者の同胞
- 患者の同胞のリスクは両親の遺伝状況に左右される。
- もし発端者の両親(の一方)が罹患しているならば、同胞のリスクは50%である。
- NF2患者の両親のいずれもが無症状であるならば、症状発現時の年齢は家族内で比較的一定なので、患者の同胞がNF2に罹患している可能性はきわめて低い。しかし臨床的に問題のない両親の生殖細胞モザイクは1例の報告があるのみである。さらに体細胞モザイク(生殖細胞モザイクを含む)はNF2散発患者の25-33%でみられる。
発端者の子
NF2患者の子はそれぞれ最大50%の確率でNF2遺伝子変異を受け継ぐ。
- もし発端者の他に罹患した家族があるならば、発端者の子がNF2変異を受け継ぐ確率は50%である。
- もし発端者が家系内でただ1人の罹患者であるならば、2つの可能性が考えられる。
- 発端者はNF2遺伝子変異の体細胞モザイクを有している。モザイクの患者の子がNF2変異を受け継ぐ確立は50%より低くなる。
- 発端者はNF2遺伝子の新生突然変異を有している。(つまり受胎期に卵子もしくは精子に存在している)新生突然変異の患者の子はそれぞれ50%の確率で変異を受け継ぐ。
- 体細胞モザイクと両側性前庭神経鞘腫を有する患者が罹患した子を持つ確率は50%より低い。もし変異が多発性腫瘍DNAには認められるが白血球DNAでは認められない場合、子のリスクはおそらく5%未満である。
他の家族
他の家族のリスクは発端者の親の遺伝的状況に左右される。もし親が罹患しているのであれば、家族構成によるが、その親の家族もリスクを有していることになる。
遺伝カウンセリングに関連した問題
リスクある親族の早期診断・治療のための検査に関する情報は、臨床的マネジメントにあるリスクのある親族に対する検査を参照
家族計画
- 遺伝学的リスクや出生前検査の可否などについての議論は妊娠前に行うのが望ましい。
- 罹患しているもしくはリスクがある若い成人に対して遺伝カウンセリング(子のリスクや生殖の選択肢を含め)が行われることが望ましい。
明らかな新生突然変異を認める家系について
常染色体優性状態の発端者の両親が非罹患者である場合には、代理父や母(例 生殖補助医療による)や、明らかにされていない養子縁組など、非医学的な方法も検討される可能性もある。リスクある無症状の家族に対する検査
リスクある若年の家族に対してNF2分子遺伝学的検査を検討することは適切である。(Surveillanceを参照)リスクある家系内保因者を早期に同定するために用いられる分子遺伝学的検査は変異解析か連鎖解析である。変異解析は、家族内の罹患者にNF2遺伝子変異がすでに明らかにされているならば、リスクある親族に対してのみ行うことができる。連鎖解析は、家族性NF2変異が確認されていない場合、家族内に罹患者が1名以上いる家系に推奨されて用いられる方法である。
早期に保因者を発見することは医学的マネジメントに影響するので、18歳未満のリスクはあるが無症状の若年者に対して検査を行うことは有益である。両親は遺伝子変異を受け継いでいない子に対する不必要な検査を避けるため、しばしば最初のスクリーニング検査が行われる年齢以前に子どもの遺伝学的状況を知りたがる。遺伝子検査の前に親子に対する教育を行うなど特別な配慮が求められる。検査結果は親と子の両方に伝えられるという方法で計画がなされるべきである。
DNAバンクは(主に白血球から調製した)DNAを将来の使用のために保存しておくものである。検査法や遺伝子、変異あるいは疾患に対するわれわれの理解が将来的に進歩するかもしれないので、罹患者のDNAをDNAバンクに提供することは考慮されるべきことである。
このサービスを行っている機関についてはDNA bankingの項を参照のこと。
出生前診断
NF2のリスクが50%の妊娠における出生前診断は、通常、妊娠約15-18週に行われる羊水穿刺か10-12週に行われる絨毛生検(CVS)で得られる胎児の細胞から抽出したDNA解析により可能である。 出生前検査が行われる前には罹患者家族においてNF2変異アレルが同定されるか、もしくは連鎖が家族内で確認されなければならない。
注:妊娠期間は最終月経の開始日あるいは超音波検査による測定に基づいて計算される。
着床前診断 はNF2変異が明らかとなっている家系では利用可能である。提供施設に関してはPGDを参照
注:GeneTestsLaboratoryDirectoryに記載されている施設で実施可能な臨床用途を含むことがGeneReviewの方針である。しかし必ずしも著者、編集者、評価者による推奨を反映するものるものではない。
更新履歴
- Gene Review著者: Mia MacCollin , MD
日本語訳者 :櫻井晃洋(信州大学医学部附属病院遺伝子診療部)
Gene Review 最終更新日: 2006.6.6. 本語訳最終更新日: 2007.5.14 - Gene Review著者: D Gareth Evans、 MD、 FRCP
日本語訳者: 小川千穂(ボランティア翻訳者),櫻井晃洋(信州大学医学部附属病院遺伝子診療部)
Gene Review 最終更新日: 2011.8.18. 日本語訳最終更新日: 2012.1.2 [in present]