Silver-Russell(シルバー・ラッセル)症候群
(Silver-Russell Syndrome)
[Synonyms:Russell-Silver Syndrome]
Gene Reviews著者: Howard M Saal, MD, Madeleine D Harbison, MD, and Irene Netchine, MD, PhD.
日本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2019.10.21. 日本語訳最終更新日: 2024.4.14.
要約
疾患の特徴
Silver-Russell症候群(SRS)は、典型的には、胎生期の非対称な成長抑制により、患児が在胎不当過小児として生まれ、出生時の相対的大頭症(出生時身長・体重に対して頭囲が+1.5SD以上の状態)、前頭部の突出を伴う目立つ前額部に加え、しばしば体の非対称を呈することを特徴とする。そして出生後は、発育不良や、時に進行性の四肢長差や摂食障害を示す。その他の臨床所見としては、逆三角形の顔、第5指の彎指、尖ったオトガイを伴う小下顎症がある。四肢の長さの成長は非対称を示すものの、その他の部分の成長障害は均衡的で、頭部の成長は正常である。治療による介入を受けていない罹患者の成人期の平均身長は、平均値に対し、-3.1±1.4SDを示す。Netchine-Harbison臨床的点数化システム(NH-CSS)は、感度に優れた点数化法である。NH-CSSの臨床的基準の4つ以上、具体的には、目立つ前額部/前頭部の突出、出生時の相対的大頭症に加えて、その他2項目を満たし、なおかつ他の疾患の可能性を除外できる罹患者については、Silver-Russell症候群という臨床診断を下すことができる。
診断・検査
SRSは遺伝学的異質性を有する疾患である。臨床診断の下りた罹患者の約60%は、遺伝学的検査により裏づけをとることができる。SRS罹患者の35%-50%は、11p15.5にあるインプリンティング制御領域1(ICR1)の低メチル化により発症する。SRS罹患者の7%-10%は7番染色体の母性片親性ダイソミー(mUPD7)が原因である。また、SRS罹患者の少数は、11p15.5にあるインプリンティングセンターの重複・欠失・転座や、7番染色体の重複・欠失・転座により生じる。稀ながら、CDKN1C、IGF2、PLAG1、HMGA2に病的バリアントをもつ罹患者も報告されている。ただ、SRSの臨床診断基準としてのNH-CSSに合致する罹患者の約40%については、分子検査、細胞遺伝学的検査で異常を確認することができない。
臨床的マネジメント
症状に対する治療:
罹患者に理想的な医療を提供するには、多職種医療チームによる追跡評価と、早期からの各科の介入が必要である。治療には成長ホルモン投与療法も含まれる。低血糖に対する予防と積極的管理が必要である。摂食障害に対する対策としては、栄養剤・カロリー補給剤の使用、胃食道逆流症に対する投薬治療、口腔の運動機能障害や食物嫌悪に対する治療、食欲増進のためのシプロヘプタジンの投与があり、必要に応じて経腸チューブ栄養も考慮する。2cmを超える脚長差については、治療が必要となる。その際、年長の子どもについては、多くの場合、骨延長術が推奨される。発達の遅れに対しては、個別の改善計画に基づいた理学療法・作業療法・言語治療が行われる。心理社会的、身体イメージの問題に対する対策としては、心理カウンセリングが行われる。重度の小下顎症や口蓋裂に対しては、頭蓋顔面分野の多職種共同医療チームでの対応が必要となる。停留精巣や尿道下裂を有する男性に対しては、泌尿器科医への紹介が必要となる。小陰茎を伴う男性、ならびに尿路・内性器奇形を伴う女性に対しては、性発達センターの多職種共同医療チームへの紹介が有益である。
定期的追跡評価 :
以下の諸点についてモニターしていく。
- 成長曲線
- 乳児(必要であれば年長の小児も)については、低血糖症を念頭に血糖値と尿中ケトン
- 幼児期の小児科健診で、成長の非対称を察知するための四肢長の評価
- 脊柱側彎の評価、中枢性思春期早発症の徴候、歯の叢生や咬合異常、言語発達
避けるべき薬剤/環境:
- 低血糖のリスクがあるため、乳幼児については長時間の空腹状態の回避
- 低血糖・低体温・治癒困難・挿管困難のリスクがあるため、待機的手術の可能な範囲での回避
遺伝カウンセリング
SRSには多数の病因があるが、概して再発リスクは低い。大多数の家系では、Silver-Russell症候群(SRS)の発端者は孤発例(家系内で1人だけの罹患者)で、見かけ上de novoのエピジェネティックあるいはジェネティックな変化(例えば、11p15に座位するICR1 H19/IGF2インプリンティングセンター1の父性メチル化の異常、あるいは7番染色体の母性片親性ダイソミー)の結果として生じたSRSである。ただ、ジェネティックな変化の本態や、伝達者である親の性別によっては、最大50%の再発リスクをもつジェネティックな変化(例えば、7番・11番染色体のコピー数バリアントや、CDKN1C、IGF2、PLAG2、HMGA2の遺伝子内病的バリアント)に起因してSRSが生じることもありうる。したがって、SRSの再発リスクを正確に評価する上では、発端者の有する発症原因としての遺伝学的メカニズムの同定が必要である。
診断
Silver-Russell症候群(SRS)は遺伝学的異質性を有する疾患である。SRSの臨床診断を下すにあたっては、「本疾患を示唆する所見」の項に載せたNetchine-Harbison臨床的点数化システム(NH-CSS)[Azziら2015]の各項目の基準を満たす必要がある。この点数化システムは、SRSと診断するにはさらなる評価が必要と判断すべき症例を特定する上でも有用なものとして受け入れられている[Wakelingら2017]。
本疾患を示唆する所見
以下のNH-CSS 臨床基準[Azziら2015]を満たす症例については、Silver-Russell症候群(SRS)を疑う必要がある。
- 在胎不当過小児(在胎週齢の平均に対して出生時体重ないし出生時身長が-2SD以下)。
- 出生後の発育不良(24ヵ月時点で、身長・体重が平均の-2SD以下)。
- 出生時における相対的大頭症(出生時体重・出生時身長に対し、頭囲が1.5SDを超えて大きい)。
- 前頭部の突出、ないし目立つ前額部(幼児[1-3歳]において、側面観で顔面平面を超えて前額部が突出している)。
- 身体の非対称(0.5cm以上の四肢長差、あるいは0.5cm未満でも他の身体各部に2ヵ所以上の非対称がみられること)。
- 摂食障害がある、もしくは24ヵ月時点でのボディマス指数が-2SD以下であること、現に栄養チューブや食欲増進のためのシプロヘプタジンを使用していること。
上記6つの基準のうちの4つを満たす罹患者については、臨床診断としてSRSが疑われるため、分子遺伝学的検査に進んでよい。稀ながら、3つしか基準を満たしていない罹患者であっても、分子遺伝学的検査でSRS陽性の結果が出る例がある。
診断の確定
SRSの診断は、Netchine-Harbisonの臨床診断基準の6項目中4項目を満たす発端者、ならびに、分子遺伝学的検査にて、11p15.5の低メチル化、7番染色体の母性片親性ダイソミー(UPD)のいずれかが確認された発端者において確定する(表1参照)。
11p15.5のインプリンティングクラスター
11p11.5の2つのインプリンティングドメインにおける遺伝子転写の制御異常に関連して生じるタイプのSRSがある。この領域の遺伝子発現制御の詳細については、「分子レベルの病原」の項を参照されたい。
7番染色体の母性片親性ダイソミー
母性UPD7が生じるメカニズムにはいくつかのものがある。「分子レベルの病原」の項を参照されたい。
SRSの診査・診断過程のためのアルゴリズムが、国際エキスパートコンセンサスから公表されている[Wakelingら2017]。図1を参照されたい。
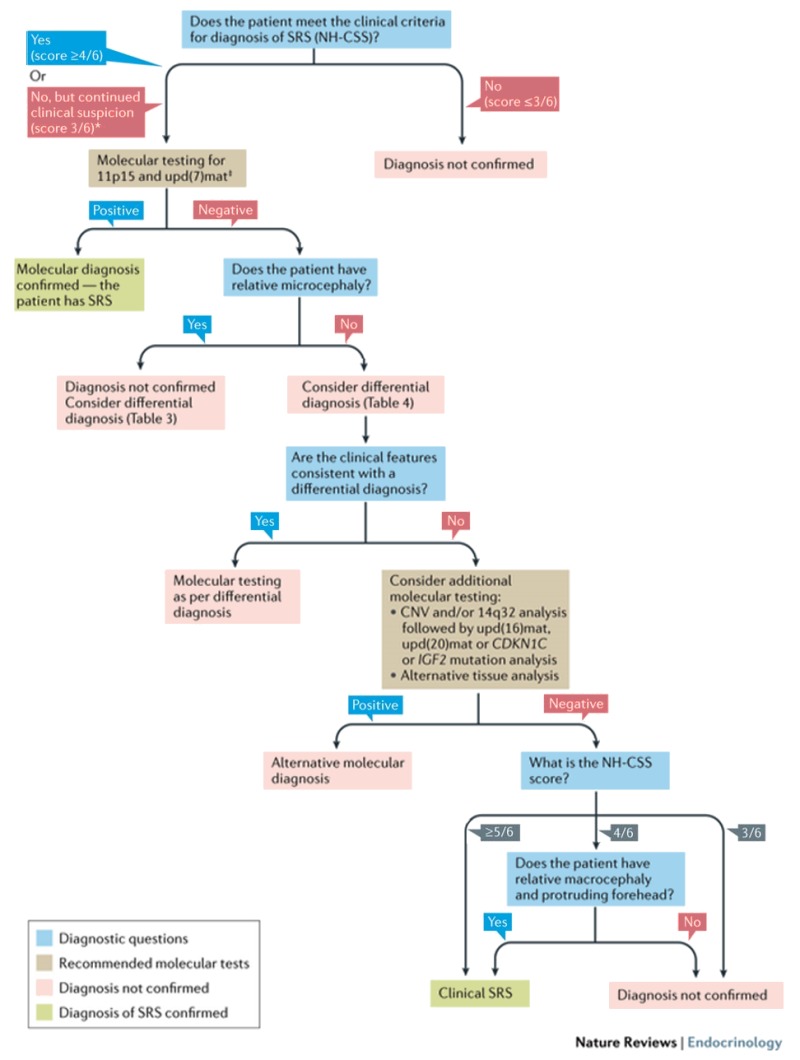
図1: Silver-Russell症候群の診査・診断のためのフローチャート
この図(原著論文にある説明文は割愛している)はWakelingら[2017]の論文に収載されたものである。この図は、クリエイティブコモンズ【表示4.0国際】ライセンスのもとに公表されている。
SRS診断のための検査は、次の順序で進めることが推奨されている。
- 11p15.5 ICR1/ICR2のメチル化解析と、母性UPD7の検査を同時並行で行う。
注:(1)異常が検出されるかどうかは、疾患の発生メカニズムと使用した検査法の関係によって決まる(表1参照)。
(2)モザイクの報告がみられる。したがって、リンパ球の検査で異常なしという結果が出た場合は、他の組織(頰粘膜細胞もしくは線維芽細胞)の検査も検討に値する。
- 11p15.5 ICR1/ICR2のメチル化解析と、母性UPD7の検査で異常が検出されなかった場合は、IGF2、CDKN1C、PLAG1、HMGA2の配列分析を考慮する。
注:最終的にSRSと診断される罹患者の中には、出生時の有意な発育不全やその後の言語発達等の遅延を理由に、SNP染色体マイクロアレイ解析(CMA)が実施されている例もある。この検査は、SRSが疑われる罹患者に対して最初に行う検査として推奨されるようなものではないが、これにより7番、11番染色体の異常が発見され、診断の確定に至る例も存在する(表1参照)。
マルチ遺伝子パネル検査
11p11.5 ICR1/ICR2のメチル化解析と、母性UPD7の検査で異常が検出されなかった場合は、IGF2、CDKN1C、PLAG1、HMGA2、ならびにその他の関連遺伝子(「鑑別診断」の項を参照)の配列解析を含めたマルチ遺伝子パネル検査を考慮すべきであろう。
注:(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。したがって、臨床医には、どのマルチ遺伝子パネルを用いれば、現状の表現型と直接関係のない意義不明バリアントや病的バリアントの検出を抑えつつ、安価に本疾患の原因遺伝子を同定できる可能性が高いかという点を判断することが求められる。
(3)検査機関によって、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエキソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法の中に、配列解析、欠失/重複解析、その他の非配列解析型検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝学的検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
注
Netchine-Harbison点数化システムで6項目中4項目を満たしたため、分子検査に回った罹患者の約40%は、陰性の検査結果を示す。こうしたグループについては、次の2項を満たすことを条件に、SRSの臨床診断を確定することが可能である[Wakelingら2017]。
(a)条件を満たした4つの診断基準のうちの2つが、目立つ前額部/前頭骨の突出と、出生時における相対的大頭症であること。
(b)他の疾患である可能性が排除されていること(「鑑別診断」の項を参照)。
表1:Silver-Russell症候群で用いられる分子遺伝学的検査
| 手法 | 検出される病的バリアント/変化 | RSの中で検出されるものの割合1 | |
|---|---|---|---|
| メチル化解析2 | 11番染色体 | H19/IGF2メチル化障害(父性)3 | 最大35%-50%4 |
| 母性UPD11の体細胞モザイク5,6 | 稀 | ||
| 11p15.5の重複(母性) | 不明7,8 | ||
| 7番染色体 | 母性UPD79 | 最大7%-10%10 | |
| 微小欠失,微小重複,7トリソミーモザイク | 稀11 | ||
| マイクロアレイ (SNPベース) |
11番染色体 | 11p15.5の重複(母性) | 不明7,8 |
| 母性UPD11の体細胞モザイク5,6 | 稀12 | ||
| 7番染色体 | 7pの微小欠失ないし微小重複,7トリソミーモザイク | 稀11,13,14 | |
| 母性UPD7 | 母性アイソダイソミー7のみ15 | ||
| STR*マーカー解析 | 11番染色体 | 母性UPD11の体細胞モザイク5,6 | 稀 |
| 7番染色体 | 母性UPD79 | 最大7%-10%10 | |
| 核型 | 11p15.5の逆位もしくは転座 | 稀5,6 | |
| 配列解析16/遺伝子標的型 欠失/重複解析17 |
CDKN1C(母性継承) | 1家系の報告18 | |
| IGF2(父性継承) | 数例の報告19 | ||
| PLAG1 | 稀20 | ||
| HMGA2 | 稀21 | ||
| 不明 | 30%-40%22 | ||
*訳注:STRはshort-tandem repeatの略。
- 検出されているバリアント/変化に関する情報については、「分子遺伝学」の項を参照されたい。
- メチル化感受性MLPA法(MS-MLPA)、メチル化感受性定量的PCR法(MS-qPCR)、サザンブロット法(主として従来法)などの、メチル化感受性を高めるために開発されたアッセイを用いることで、11p15.5のエピゲノムやゲノムの変化を検出することができる。メチル化感受性アッセイにより、DNAのメチル化の変化、微小欠失と微小重複、片親性ダイソミー(UPD)に起因するSRSの診断が可能である。メチル化に関するデータの解釈にあたっては、コピー数検査の結果を頭に入れておく必要がある。なぜならば、両親の遺伝子量の相対的寄与割合を変化させるようなコピー数バリアント(例えば、父性重複)が、メチル化状態の異常に関係していることがあるからである。欠失や重複の切断点を明らかにするため、MLPA法の検査に続いてマイクロアレイ検査を行うことがあることに注意されたい。11p15.5の母性UPDを確認するその他の手法としては、short-tandem repeat(STR)解析、SNP解析などがある[Kerenら2013]。
- 少数ながら、H19ないしIGF2のみの低メチル化を示す罹患者も存在する[Bartholdiら2009]。
- 11p15.5の低メチル化は受精後に生じる。そのため、モザイクに起因する偽陰性が生じうる。他の組織(例えば頰粘膜の細胞や線維芽細胞)を用いた検査を併せて行うべきである。
- Bullmanら[2008]
- Lukら[2016a]
- Fisherら[2002],Eggermannら[2005],Schönherrら[2007]
- Heideら[2018]
- アイソダイソミー、ヘテロダイソミーの両方[Bernardら1999,Priceら1999]、ならびに分節的母性UPD[Hannulaら2001,Eggermann 2008]が報告されている。母性UPD7その他の7番染色体再構成の例については、モザイクの存在が知られている。したがって、別の組織を用いて検査を行うことが適切であろう[Reboulら2006]。
- Hannulaら[2001],Kimら[2005]
- Courtensら[2005],Floriら[2005],Font-Montgomeryら[2005]
- Lukら[2016b]が、UPD11を有するSRS罹患者28人から成る1群を報告している。
- GRB10、GFBP1、GFBP3を含む母性アレルの7p11.2-p13のde novoの欠失が報告されている[Monkら2000]。
- MESTを含む父性アレルの7q32.2のde novoの欠失が報告されている[Carreraら2016]。
- 注:SNPアレイ解析では、母性UPDの中のアイソダイソミーのみ検出可能である。母性UPDの28.8%(SRS全体の2%-3%)がアイソダイソミーである[Chantot-Bastaraudら2017]。
- 配列解析を行うことで、benign、likely benign、VUS、likely pathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、遺伝子内の小欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、遺伝子の部分欠失や部分重複、1遺伝子全体の欠失や重複、複数遺伝子の欠失や重複は検出されない。配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
- 遺伝子標的型の欠失/重複解析では、遺伝子内の欠失や重複が検出される。具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失ないし重複の検出を目的に設計された遺伝子標的型マイクロアレイなどがある。
- CDKN1Cの機能獲得型バリアントの分離された1家系4世代の例が報告されている。
- SRSと父性継承のIGF2の機能喪失型バリアントをもつ1家系がBegemannら[2015]により報告されている。また、Tümerら[2018]による別の数家系のレビューがある。
- PLAG1の機能喪失型バリアントは、家族性の2例、孤発性の1例で報告されている[Abi Habibら2018]。
- HMGA2の機能喪失型バリアントは、孤発性の2例で報告されている[Abi Habibら2018]。
- Netchine-Harbison点数化システムで6点中4点が確認されて分子検査に移行した罹患者の約40%については、検査によって診断確定に至らない。「注」の項目を参照されたい。
臨床的特徴
臨床像
Silver-Russell症候群(SRS)の典型的所見は、妊娠中の非対称な成長抑制の結果として生じる相対的大頭症(出生時の身長・体重に対して+1.5SD以上を示す頭囲)を伴う出生時の在胎不当過小、前頭骨の突出を伴うことの多い目立つ前額部、しばしばみられる体の非対称である。その後、出生後の発育不全や、症例によっては進行性の四肢長差、満1歳に達するまでの間の重度の摂食障害へと続いていく。その他の所見としては、逆三角形の顔、第5指の彎指、細いオトガイを伴う小下顎症がある。四肢長の非対称の問題は別として、発育障害は均衡的に推移し、頭の成長は正常である。治療を行わなかった場合の成人の平均身長は、平均に対して-3.1±1.4SDの値となる。
SRSは、病因の上で異質性を有する。SRSの診断・管理に関する国際エキスパートコンセンサスの最近の発表[Wakelingら2017]においては、Netchine-Harbison臨床的点数化システム[Netchineら2007,Azziら2015]の感度が他の点数化システムに抜きん出ているとして、これが採用されている。
Netchine-Harbison臨床的点数化システム(NH-CSS)[Azziら2015]とは、以下のようなものである。
- 在胎不当過小(在胎週齢の平均に対して出生時体重ないし出生時身長が-2SD以下)。
- 出生後の発育不全(24ヵ月時点で、身長・体重が平均の-2SD以下)。
- 出生時における相対的大頭症(出生時体重・出生時身長に対し、頭囲が1.5SDを超えて大きい)。
- 前頭骨の突出、ないし目立つ前額部(幼児[1-3歳]において、側面観で顔面平面を超えて前額部が突出している)。
- 身体の非対称(0.5cm以上の四肢長差、あるいは0.5cm未満でも他の身体各部に2ヵ所以上の非対称がみられること)。
- 摂食障害があること、もしくは24ヵ月時点でのボディマス指数が-2SD以下であること、もしくは現に栄養チューブや食欲増進のためのシプロヘプタジンを使用していること。
SRSの臨床診断は、出生時の相対的大頭症と前頭骨の突出を含め、乳児が4つ以上の条件を満たしている場合に下される。それ以降の年齢の小児や成人については、顔面所見は変化し、前頭骨の突出も消失している可能性がある。したがって、NH-CSSを年長の小児や成人に適用するにあたっては、前頭部の突出の評価を小児期のうちに行うとともに、2歳前後に撮った側貌写真で行う評価する必要がある。
SRSの罹患者であれば、それを追認するような以下のような所見がみられるはずである。
- 大泉門閉鎖の遷延
- 逆三角形の顔
- 小下顎症
- 歯の叢生
- 下がった口角
- 甲高い声
- 筋組織量の減少
- 肩の窪み
- 肘関節の低形成
- 第5指の彎指もしくは短指
- 脊柱側彎
- 発汗過剰
- 空腹時低血糖
- 言語発達の遅れ
- 運動発達遅滞
- 尿路性器奇形
成長
SRSで最も早くからみられる所見は成長の異常である。大多数の患児は在胎不当過小で生まれ、身長・体重は平均に対して-2SD以下を示す。成長速度に関しては正常範囲内にあることもあるが、SRSの罹患者については有意な追いつき成長を示すことはほとんどない。両親が高身長の場合を除き、2歳の段階で大多数の患児の身長は平均に対して-2SD以下の値を示す。ヨーロッパのSRS患児については、成長曲線のデータが公表されている[Wollmannら1995]。Wollmannのデータと重ね合わせて描いた北アメリカの子どもの成長曲線が、MAGIC財団を通じて入手可能である(2020年5月22日アクセス済)。
未治療のSRS罹患者を資料として用いたヨーロッパの2つの研究では、平均身長に対して男性は-3.7から-3.5SD、女性は-4.2から-2.5SDの範囲を示したという[Wollmannら1995,Binderら2013]。四肢長の非対称を除けば、発育障害は均衡的に生じ、頭部の成長は正常である。
SRSであろうとの臨床診断を受けた子どもで、その後、追いつき成長を示した例については、その大半が典型的SRS以外の疾患であったという報告がある[Saalら1985]。
SRS患児の成長をコントロールするための成長ホルモン治療の施行に関しては、「臨床的マネジメント」の項を参照のこと。
注:SRS患児の多くは、ヒト成長ホルモンの投与を受けたとしても、思春期における骨年齢の急激な進行をうまくコントロールできない限り、最終的に正常の身長を達成するには至らない。
他の内分泌の問題
他の内分泌の問題としては、早発副腎皮質性第二次性徴、思春期早発症、インスリン抵抗性があり、これらの影響により、成長ホルモン治療後も最終身長は目標に届かない。
骨格の異常
骨格の異常としては、
- 患側の成長が障害される片側劣成長による四肢長の非対称
- 第5指の彎指ないし短指。SRS罹患者では、これが最も頻発する骨格奇形(小奇形ではあるが)である。
- 脊柱側彎。ある研究によると、SRS罹患者の36%にこれがみられるという[Abrahamら2004]。より近年の研究では、脊柱側彎ないし脊柱後彎が21%にみられ、18%については矯正術が必要であったという[Yamaguchiら2015]。
神経発達
両親にとっては、成長の問題とは別に、神経発達の問題も大きな懸念材料である。SRSの子どもは、発達遅滞(運動発達と認知機能の発達の両面の遅延)や学習障害に関して高リスク状態にあるとの研究結果が出ている。
11p15のメチル化異常と母性UPD7の両方を含むSRS児のコホートを対象とした大規模なレビューでは、発達遅滞が患児の34%にみられ、その大多数は軽度遅延であったとされている。発達遅滞は、UPD7患児のほうが11p15メチル化異常患児より多くみられたという(65%対20%)。言語発達の遅れは、両群ともに多くみられている[Wakelingら2010]。認知機能に関する正確な予後を把握するためには、分子検査での確定診断がなされたSRS患児に対して早期から適切な医療を行った研究をさらに進めていく必要がある
摂食障害と低血糖症
SRSの子どもは、皮下脂肪がほとんどなく、しばしば低食欲・口腔運動機能障害・摂食障害を示し[Fukeら2013]、特に長時間の空腹で低血糖を起こすリスクを有する[Wakelingら2017]。SRS患児について低血糖の要因を調べた研究で、カロリー摂取不足(多くは食欲と摂食の低下に起因して続発性に生じるもの)、ボディマスの低下、ならびに数人の子どもにみられた成長ホルモン分泌低下などがその要因として挙げられている[Azcona & Stanhope 2005]。大多数の患児には低血糖に伴う臨床症状がみられたが、無症状の例も数例みられた。
発汗
幼児期の発汗の背景に低血糖が関与していることがあるものの、SRS患児については、低血糖がなくても発汗がみられることがしばしばある[Stanhopeら1998]。
消化器疾患
消化器疾患が多くみられ、その内容としては、胃食道逆流症、食道炎、食物嫌悪、嘔吐、便秘、発育不良がある。1つの大規模研究にて、消化器系の問題が77%の患児で報告されている。55%の患児は重度の胃食道逆流症を示すが、SRS患児の場合は嘔吐を伴わない非典型的な胃食道逆流症を示すこともある[Marsaudら2015]。食物嫌悪や誤嚥を示す患児に対しては逆流性食道炎を疑う必要がある。
頭蓋顔面奇形
頭蓋顔面奇形も多くみられる。SRS罹患者の中にはPierre Robin sequenceや口蓋裂を伴う例もみられる。Wakelingら[2010]は、SRS罹患者群において、11p15.5メチル化異常をもつ罹患者の7%に口蓋裂ないし口蓋垂裂を認めた一方で、母性UPD7の罹患者にこれらはみられなかったとしている。Pierre Robin sequenceを伴う罹患者については、閉塞性無呼吸の監視が必要である。
歯や口腔の異常も多くみられる。矮小歯、高口蓋、相対的小下顎・小口に伴い二次的に生じる歯の叢生などが報告されている[Orbakら2005,Wakelingら2010]。口腔顔面所見の中で最も多くみられるものは、過蓋咬合と叢生である[Hodgeら2015]。
叢生下で口腔衛生不良があると、齲蝕の危険が高まる。
尿路性器奇形
尿路性器奇形の報告もみられる。最も多くみられる奇形は、男性における尿道下裂と停留精巣である[Bruceら2009,Wakelingら2017]。女性では、Mayer-Rokitansky-Kuster-Hauser症候群(見た目は正常な外性器ながら、膣や子宮が低形成ないし欠損を示す)の報告がみられる[Bruceら2009,Abrahamら2015]。腎奇形はあまり多くないものの、馬蹄腎と異形成腎の報告がみられる[Wakelingら2010]。
心奇形
心奇形はそれほど多くないものの、大規模研究[Wakelingら2010]、小規模研究[Ghanimら2013]の両方で報告されている。心奇形の発生頻度は5.5%程度と思われる[Ghanimら2013]。
遺伝型-表現型相関
Bruceら[2009]は、メチル化感受性制限酵素であるHpaⅡとNotⅠを用いてH19のメチル化の程度を計測し、H19の極度の低メチル化、H19の中等度の低メチル化、H19の正常なメチル化、母性UPD7(H19のメチル化は正常)から成るスケールを開発した。その結果、H19の極度の低メチル化(平均の-6SD以下、もしくは9%未満のメチル化)を呈するSRS患児は、中等度の低メチル化を示すSRS患児や母性UPD7のSRS患児に比べ、より重度の骨格異常(橈骨上腕骨関節の脱臼、合指趾、大きな四肢非対称、脊柱側彎など)を示す傾向にあったという。
Wakelingら[2010]は、11p15.5 ICR1 IGF2/H19メチル化異常に起因するSRS患児の臨床所見を、母性UPD7の患児と比較している。その結果、11p15.5 ICR1の低メチル化群のほうが、母性UPD7群より第5指の彎指や先天奇形の頻度が高い一方、学習障害や言語障害については、母性UPD7群のほうがICR1低メチル化群より頻度が高かった。
成長ホルモン治療に対しては、母性UPD7のSRS患児のほうが11p15.5エピ変異の患児より身長の増加量が大きかった。これはおそらく、11p15.5メチル化異常を伴う患児の場合、インスリン様成長因子Ⅰ(IGF1の遺伝子産物)のレベルが上昇し、その結果、IGF1抵抗性が高まる一方、母性UPD7のSRS患児の場合は、在胎不当過小を示す他の子どもたちと同じ反応様式を示すためであると思われる[Binderら2008]。
頻度
発生頻度は不明であるが、以前は30,000人に1人から100,000人に1人と推定されていた(Annick Toutain, Orphanet)。最近、エストニアで行われた後ろ向き研究では、SRSの最小発生頻度を15,886人に1人としている[Yakorevaら2019]。
遺伝学的に関連のある疾患(同一アレル疾患)
Silver-Russell症候群関連遺伝子の病的バリアントに起因して引き起こされる他の表現型を表2に示した。
表2:同一アレル疾患
| 分子的変化1 | 表現型1 |
|---|---|
| 11p15.5(Beckwith-Wiedemann症候群重要領域)のインプリンティングドメインにおける遺伝子転写制御の異常、もしくは母性継承性のCDKN1Cの機能喪失型病的バリアント | Beckwith-Wiedemann症候群 |
| ICR2の脱メチル化、ICR1の高メチル化2、11p15の父性UPD3を含む11p15領域の分子的変化 | 単発性の片側過形成症 |
| 父性ICR1の脱メチル化の体細胞モザイク4 | 単発性の片側低形成症 |
| ICR1の高メチル化、11p15.5の父性UPD、微小欠失・微小挿入といったゲノム異常などを含む11p15.5の構成的変化5 | 単発性のWilms腫瘍 |
| 母性遺伝性のCDKN1Cの機能獲得型病的バリアント | IMAGe症候群 |
- より詳細な情報については、ハイパーリンクのGeneReviewないし引用文献を参照のこと。
- Martinら[2005]
- Shumanら[2002]
- Zeschnigkら[2008],Eggermannら[2009]
- Scottら[2008]
鑑別診断
子宮内の発育抑制と低身長
子宮内において発育抑制・低身長をきたすあらゆる疾患が、Silver-Russell症候群(SRS)との鑑別診断の対象となる。不均衡型低身長がみられる場合は、骨系統疾患が示唆され、SRSの可能性は否定できる。SRS類似の骨系統疾患と確実に区別するには、骨格の診査が必要である。
注:SRS患児の場合は、骨年齢に遅延がみられることが多い。ただ、骨年齢の遅延は非特異的な所見であり、その他多くの子宮内発育抑制疾患の子どもにおいてもしばしばみられる。
染色体異常
染色体不均衡を伴う数多くの疾患で、在胎不当過小と出生後の発育不良がみられ、SRSと誤診されることがある。染色体マイクロアレイ解析、できればSNPベースのマイクロアレイ解析を行えれば、微小欠失・微小重複や、片親性ダイソミーをうかがわせるホモ接合領域、さらには血族結婚例でみられる稀な潜性遺伝疾患などを同定するのに役立つ[Groteら2014]。いくつかの染色体の片親性ダイソミー、例えば6番、14番(Temple症候群)、16番、20番といったところの片親性ダイソミーで、SRS類似の表現型が現れるとの報告がみられる[Sachwitzら2016,Wakelingら2017,Geoffronら2018]。
小頭症
SRS患児は、頭囲そのものは正常で、相対的大頭症の状態を呈する。明らかな小頭症がみられる場合は、別の原因を模索する必要がある。
表3に、Silver-Russell症候群との鑑別診断が必要な症候群の概要を示す。
表3:子宮内発育抑制と出生後の発育不良を呈し、Silver-Russell症候群との鑑別診断が必要となる疾患
| 疾患名 | 原因遺伝子 | 遺伝形式 | 他の臨床所見 | ||
|---|---|---|---|---|---|
| SRSと重なる所見 | SRSと異なる所見 | ||||
| 単一遺伝子疾患 | 3-M症候群 | CCDC8 CUL7 OBSL1 |
AR |
|
|
| IMAGe症候群 | CDKN1C | AD1 |
|
|
|
| Bloom症候群 | BLM | AR |
|
|
|
| Nijmegen染色体不安定症候群 | NBN | AR | カフェオレ斑 |
|
|
| Warsaw破壊症候群(OMIM 613398) | DDX11 | AR | 第5指彎指 |
|
|
| Fanconi貧血 | 20遺伝子超2 | AR AD XL |
カフェオレ斑 |
|
|
| Meier-Gorlin症候群(OMIM 224690) | CDC45 CDC6 CDT1 GMNN MCM5 ORC1 ORC4 ORC6 |
AR AD |
前頭部の突出 |
|
|
| インスリン様成長因子不応症(15q26.1欠失を含む)3 | IGF1R | AR AD |
|
|
|
| 染色体異常 | 2倍体/3倍体の混数倍数体4 | 四肢非対称 |
|
||
| Temple症候群(母性UPD14,父性14番染色体欠失ないし14q32脱メチル化)(OMIM 616222) | SRSと多数の所見の重なりあり |
|
|||
| 催奇形性疾患 | 胎児性アルコール症候群 |
|
|
||
AD=常染色体顕性;AR=常染色体潜性;XL=X連鎖性
- IMAGe症候群を引き起こすCDKN1Cの病的バリアントは、典型的には常染色体顕性遺伝の形で継承される。
ただし、病的バリアントが母性伝達された場合のみIMAGe症候群に至る。 - Fanconi貧血には、BRCA2、BRIP1、ERCC4、FANCA、FANCB、FANCC、FANCD、FANCE、FANCF、FANCG、FANCI、FANCL、FANCM、MAD2L2、PALB2、RAD51、RD51C、RFWD3、SLX4、UBE2T、XRCC2といった遺伝子の病的バリアントが関与する。
- Bruceら[2009],Ocaranzaら[2017]
- Grahamら[1981]
- Ioannidesら[2014],Kagamiら[2015],Geoffronら[2018]
臨床的マネジメント
最初の診断に続いて行う評価
Silver-Russell症候群(SRS)と診断された罹患者については、疾患の範囲を確定するため、まだ行っていないようであれば、次のような評価を行うことが推奨される。
- 成長曲線の評価と描記。これについてはMAGIC財団のホームページを参照されたい。
- 四肢長の非対称、尿路性器奇形、脊柱側彎、思春期早発症、口腔や頭蓋顔面の異常を評価するための診査
- 小児内分泌内科医への紹介
- 小児消化器内科医や栄養士への紹介
- 胃食道逆流症(GERD)の疑いがある子どもに対して、ビデオ嚥下検査・胃排出能検査・pH検査・内視鏡検査などで食道炎の評価を行う。
- SRS罹患者に腸回転異常の報告があることから、摂食障害・便秘・胃排出能の低下がみられる患児に対しては、腸回転異常の可能性を排除しておく必要がある。
- 神経認知発達、言語発達、筋緊張の状態についてのスクリーニング評価
- 臨床遺伝医や遺伝カウンセラーとの面談
症候に対する治療
SRSは、多岐にわたる肉体的、機能的異常を惹起する。SRS罹患者に対して理想的な管理を行うには、多職種連携医療に基づく追跡と、早期からのSRSに特化した医療の提供が必要である。多職種連携医療チームは、それぞれ小児分野を専門とする内分泌医、消化器医、栄養士、臨床遺伝医・遺伝カウンセラー、頭蓋顔面チーム、整形外科医、神経科医、言語聴覚士、心理士で構成されるのが望ましい。
成長、ならびに成長ホルモン治療
他者と異なる身体や低身長を呈する疾患の子どもは、ボディイメージに敏感であることが多い。こうした身体的要素は、自己イメージ、仲間との関係、社会性といった点に関して、重要な役割を果たすことが考えられる。そのため、SRS患児については、早期に小児内分泌科医に紹介することが重要である。
- 成長ホルモン治療
原因の如何を問わず、子宮内発育抑制を有する子どもに対する成長ホルモン治療は、成長や最終身長に関して有意な改善をもたらす。追いつき成長を示さない子どもたちに対しては、この治療が特に効果的である[Dahlgrenら2005,Jensenら2014,Zanelli & Rogol 2018]。SRS患児については、成長ホルモン投与の効果が特に大きいという[Toumbaら2010,Binderら2013]。
- Smeetsら[2016]は、成長ホルモン治療を行ったSRS患児について、SRSを有しない小児と同じ程度の身長の伸びを獲得したものの、SRS患児のほうが治療開始時点での身長が低かったため、最終身長についてはSRS群のほうが低かったとしている[Smeetsら2016]
- 成長ホルモン治療の目標は、成長速度の改善は当然のことであるが、それ以外にも、身体構成(特に痩せ状態)・精神運動発達・食欲の改善、低血糖リスクの低減、理想的な線形成長の達成といったこともその目標となる[Wakelingら2017]。
- Rizzoら[2001]は、成長ホルモン治療を行ったSRS患児は、有意な身長の伸びを示したものの、体幹や四肢の非対称については変化がみられなかったとしている。
- こうした治療は、成長障害の管理に関して経験豊富な医療機関で行われることが望ましい。
- 成長ホルモン分泌不全
成長ホルモン治療は、ふつう、成長ホルモン分泌不全が確認されることがその条件とされる。しかし、SRSにおいては、成長ホルモン分泌不全はあまりみられない[Wakelingら2017]。
- SRSにおいては、成長ホルモン分泌不全の有無にかかわらず、成長ホルモン治療が適応となる。
- 絶食を伴う成長ホルモン分泌不全の検査は、低血糖を誘発するリスクがあることから禁忌である[Wakelingら2017]。
- 骨年齢の進行状況と思春期
SRS罹患者については、副腎皮質性思春期早発症、かなり早期から加速度的に現れる中枢性思春期早発症、インスリン抵抗性といった点に関する徴候の監視が非常に重要である。中枢性思春期早発症(女性で12歳、男性で13歳以前)が確認された子どもに対しては、正常な成人期の身長を確保する目的で、ゴナドトロピン放出ホルモン(GnRH)アナログ製剤を用いた個別治療も検討に値する[Wakelingら2017]。
低血糖
低血糖の予防が必要で、もし低血糖がみられる場合は、それに対する積極的な治療が必要となる。
- 空腹状態が長引いた後(夜間を通じて眠るようになった乳児を含む)、過剰な運動の後、ならびに病気のときなどは、尿中ケトンレベルの監視を行うこと。
- 摂食を頻回に行い、長時間の空腹を避けること(乳児の場合は4時間以内)。
- 複合炭水化物の使用。
消化器疾患
消化器疾患については、早期かつ適切に対処する必要がある。成長や栄養に関しては、乳児期に監視を始めることが肝要である。SRSの乳児は筋組織の発達が弱く、内在的な発育障害に加えて、摂食障害の問題も併せもつことが多い。消化器疾患・摂食障害・栄養不良がSRS患児の70%にみられるとの研究報告もある[Marsaudら2015]。したがって、多くの場合、積極的な摂食補助が必要になる。ただ、出生後の体重の追いつきがあまりに急速に生じると、その後、代謝性疾患のリスクが高まることにつながるため、体重増加に関しては注意深く監視することが必要である。
具体的戦略としては、以下のようなことが推奨される。
- 栄養補助やカロリー補給のためのサプリメントの使用。
- プロトンポンプ阻害薬やH-2ヒスタミンブロッカーといった薬剤を適切に使用しながら行う胃食道逆流症の治療。
- 口腔運動機能の問題ならびに食物嫌悪に対して行う言語療法や作業療法。
- 食欲刺激剤としてのシプロヘプタジンの使用。シプロヘプタジンについては、体重増加や成長速度に関する有効性が確認されている。
- 他の手法が無効に帰すような極端な症例に限られはするものの、食物嫌悪や胃食道逆流症に対して行う胃瘻・空腸瘻造設を伴う経腸栄養の使用[Wakelingら2017]。これに併せて噴門形成術を施行する場合もある。非自発的栄養補給を行うにあたっては、過度に急激で過量の体重増加を避ける必要がある。
骨格の異常
整形外科的問題として最も多くみられるのは、四肢長差である。四肢長差は、数センチに及ぶことがあり、上肢・下肢の一方に生じることも両方に生じることもある。機能的には、下肢の非対称のほうが、歩行に影響が及ぶだけに、より重大である。脚長差が2cmを超えると、代償性の脊柱側彎をきたすことがあるため、治療による介入が必要となる。治療の第一歩としては、片方の靴底の嵩上げが考えられる。年長の小児になると、骨延長術による四肢長の延長が、より一般的に選択されるようになる。1セグメントの延長だけで十分な場合は、最終身長に近い状態であれば、成長の完了を待つことなく内固定器を用いた大腿骨の延長術が広く行われる[Goldmanら2013]。比較的年少の子どもで、脚長差が4cmを超える場合は、外固定器を用いた下部セグメント(脛骨)の延長が行われる[Goldmanら2013]。
脊柱側彎や脊柱後彎も、SRSではよくみられる問題である[Yamaguchiら2015]。下肢長の非対称と脊柱側彎との関係性は明らかではないが、四肢長の非対称を有する多くの子どもたちに代償性の脊柱側彎が生じる。これに対する最初の治療としては、モニタリングとブレースの装用が選択される。ただ、多くの罹患者については、その後、矯正術が必要となる[Yamaguchiら2015]。
神経発達
- 筋緊張低下を有する乳児に対しては、早期治療計画策定や理学療法に向けた紹介。
- 発達遅滞が明らかな子どもに対しては、必要に応じ、早期治療、言語治療、作業療法・理学療法に向けた紹介。
- 学齢期の子どもに対しては、適切な神経心理学的検査、ならびに必要に応じ治療的介入も含めた個別的教育計画を通じて行う、学習障害に対応するための学校教育システムとの共同作業。
- 必要に応じ、心理社会的問題や身体イメージの問題に対応するための心理カウンセリングへの紹介
頭蓋顔面奇形
重度の小下顎症、口蓋裂、顎間関係の異常といった問題を有する子どもに対しては、多職種連携の口蓋裂チームや頭蓋顔面チームでの管理が推奨される。成長の完了した年長児に対しては、歯科的ケア、叢生を除去する矯正治療に続いて、顎顔面の手術が必要になることがある。それほど重度ではない場合は、小児歯科医や矯正歯科医の手で、通常の手法で口腔衛生や叢生の問題に十分対応することが可能である。
泌尿生殖器系の異常
停留精巣や尿道下裂に対しては、小児泌尿器科医による対応が最も望ましい。
小陰茎の男性、ならびに内的泌尿生殖器奇形を有する女性(Mayer-Rokitansky-Kuster-
Hauser症候群のことがある)に対しては、多職種連携医療を行う性発達障害(DSD)センターへの紹介が有益である。
定期的追跡評価
SRS患児のモニタリングに関するガイドラインは、SRSの診断・管理の国際合意に関する声明[Wakelingら2017]の中で明確に述べられている。
次のような形の対応が望ましい。
- 成長速度や過度の体重増加に注意を払いつつ、成長の状況を監視すること。
- ケトン性低血糖症を念頭に、尿中ケトンや血糖値の状態を監視すること。これは、乳幼児期や小児期において、大きな頭囲、痩せ型体形、食欲不振を呈する例では特に重要である。低血糖症を防止するための尿中ケトンの監視は、乳児の場合は摂食頻度が低下してきているとき、子ども全般については摂食量の低下を伴う急病時もしくは発熱時、年長の子どもについては身体活動量が増加したときに行う。
- 幼児期までは、小児科健診のたびごとに四肢長差の診査や計測を行うこと。乳児については、下肢長の計測を両脚について行って両脚の長さを記録し、成長曲線には長いほうのデータをプロットする。年長の子どもの身長計測については、腰を水平に保てるだけの適切な高さの台を短いほうの脚の下に置いて行う。
- 来院ごとに脊柱側彎の有無を診査すること。
- 進行性の副腎皮質性思春期早発症、ならびに中枢性思春期早発症を監視すること。その理由は、これらがあると、骨年齢が急速に進行して、成長ホルモン治療を長期にわたって行っても最終身長が伸びないリスクがあるからである。
- 発達遅滞、特に運動発達や言語発達の遅れに関して、慎重な監視を行うこと。
避けるべき薬剤/環境
低血糖の危険があるため、乳幼児については長時間の空腹を避ける。
待機的手術については、可能な限り避ける。手術がどうしても避けられない場合は、低血糖・低体温・創傷の難治性・挿管困難(歯列不整と小下顎により気道の視認と挿管操作が難しいことによる)といったリスクに十分気を配る。
リスクを有する血縁者の評価
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「Clinical Trials.gov」、ならびにヨーロッパの「EU Clinical Trials Register」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
大多数の家系においては、Silver-Russell症候群の発端者は孤発例(家系内で1人だけの発生例)で、見かけ上de novoと思われるジェネティックな変化もしくはエピジェネティックな変化(H19/IGF2インプリンティングセンター1における父性メチル化の欠如、もしくは7番染色体の母性片親性ダイソミー)により生じたSRSである。一方、SRSは、最大50%の再発リスクを伴うジェネティックな変化(7番・11番染色体のコピー数バリアント、もしくはCDKN1C、IGF2、PLAG1、HMGA2の遺伝子内病的バリアント)の結果として生じることもある。なお、再発リスクについては、ジェネティックな変化の本態、伝達者である親が父親か母親かで変わってくる。したがって、SRSの正確な再発リスクを算出するためには、発端者における原因となった遺伝学的メカニズムの同定が必要である。
家族構成員のリスク
表4:発端者の同胞、ならびに発端者の子への再発リスク
| 染色体/インプリンティング領域/遺伝子 | ジェネティックな変化 | 再発リスク | |
|---|---|---|---|
| 発端者の同胞 | 発端者の子 | ||
| 11p15.5ICR1 H19/IGF2領域 | 低メチル化(父性) | 低い1,2,3,4 | 低いが、経験的数値として公表されたものはない |
| 11p15.5 | 母性UPD11の体細胞モザイク | 1%未満 | 低いが、経験的数値として公表されたものはない5 |
| コピー数バリアント6 | 50%以下5,7,8 | 50% | |
| 7番染色体 | 母性UPD79 | 1%未満10 | 低い10 |
| 微小欠失,微小重複 | 保因者である親の状態により0-50% | 女性罹患者で50%,男性罹患者で0%(ただし別の表現型の異常が出現する可能性あり) | |
| 7トリソミーモザイク | 低い | 低いが、経験的数値として公表されたものはない | |
| CDKN1C | 遺伝子内の病的バリアント11 | 0%もしくは50%12 | 発端者が女性の場合は50%,発端者が男性の場合は0% |
| IGF2 | 0%もしくは50%13 | 発端者が男性の場合は50%,発端者が女性の場合は0% | |
| PLAG1 | 0%もしくは50%14 | 50% | |
| HMGA2 | |||
この表は、Eggermannら[2016]の表1を引用したもの
ICR1=インプリンティング制御領域1
UPD=片親性ダイソミー
- エピ変異例の7-25%に、多座位インプリンティング障害(MLID)が検出されることがあり、これまでに、孤発性のMLID症例で単一遺伝子の病的バリアントが報告された例がみられる。
- Bartholdiら[2009]は、ともにH19/IGF2インプリンティング制御領域1を有する父娘例を報告している。
- 同胞においてエピゲノムの病的バリアントが検出された2家系の、非罹患者の父親2人については、生殖細胞系列モザイクの可能性が高いものと考えられる[Bartholdiら2009]。
- Begemannら[2015]
- 染色体異常の種類、染色体断片のサイズ、継承の元となった親の性別といった要素により、リスクは最大で50%になることがある。
- 6.11p15.5領域を含む不均衡型相互転座や、CDKN1Cを含む11p15.5の父性微小欠失、母性微小重複といった染色体異常がある。
- Abi Habibら[2017]は、11p15.5 ICR1欠失の父性継承に起因して低メチル化が生じ、父子が罹患者となった1家系例を報告している。Abi Habibらは、ICR1の低メチル化が原因で生じたSRS罹患者の1%程度がこうしたタイプの欠失と推測されると述べている。
- Heideら[2018]は、CDKN1Cを含む11p15.5 ICR2の重複の母性継承例を報告している。
- 転座の可能性を排除するためには、核型分析を行う必要がある。
- 7番染色体の関与する家族性転座が基になった母性UPD7の1例が報告されている。染色体の転座の可能性が否定された場合、発端者の同胞や子どもへの再発リスクは低い。
- 母性伝達によるCDKN1Cの病的バリアント[Brioudeら2013]、父性伝達によるIGF2の病的バリアント、ないし、両親どちらからの伝達の可能性もあるPLAG1やHMGA2の病的バリアント[Abi Habibら2018]。
- De novoの病的バリアント、あるいは父性伝達の場合は0%、母性伝達の場合は50%。
- De novoの病的バリアント、あるいは母性伝達の場合は0%、父性伝達の場合は50%。
- De novoの病的バリアントの場合は0%、両親のいずれかからの伝達の場合は50%。
関連する遺伝カウンセリング上の諸事項
DNAバンキング
DNAバンキングとは、将来、使用できるようになった場合に備えてDNA(通常は白血球から抽出したDNA)を保存しておくことをいう。検査の手法や、遺伝子・アレルバリアント・疾患等に対するわれわれの理解が、将来はより進歩していくことが予想される。そのため、罹患者のDNAについては、保存しておくことを検討すべきである。
出生前検査ならびに着床前遺伝学的検査
出生前検査ならびに着床前遺伝学的検査
11p15.5 ICR1 H19/IGF2領域における父性の脱メチル化
これまでの研究で、絨毛採取(CVS)の行われる妊娠10-12週までの段階では、可変メチル化領域の確立がまだ完全にはなされていないことがわかっている。したがって、11p15.5 ICR1 H19/IGF2領域における父性の脱メチル化を、信頼性をもって検査することはできない[Paganiniら2015]。
さらに、羊膜細胞で可変メチル化パターンの信頼に足る検査法を確立するための研究についても、検証済のものは今のところ存在しない。また、エピゲノムの病的バリアントの大多数がモザイクであることが考えられる。そうなると、信頼に足る出生前検査はさらに困難なものになる[Eggermannら2016]。
母性片親性ダイソミー(UPD)
母性UPD7については、出生前診断が可能である。7番染色体の母性アイソダイソミーは、SNP染色体マイクロアレイ(CMA)で診断可能である。7番染色体の母性ヘテロダイソミーについては、胎児のSNP CMAを解析し、それを両親のSNP CMAと比較することで診断可能である。
染色体異常
発端者で原因となった染色体異常(11p15.5領域を含む不均衡型転座、CDKN1Cを含む11p15.5の父性の微小欠失や母性の微小重複など)が同定されているような場合は、絨毛採取もしくは羊水穿刺によって得られた胎児の細胞についてSNP CMAを行うことで、出生前検査が可能である。重複や欠失の大きさによっては、判明している染色体異常に関する着床前遺伝学的検査も可能である。
遺伝子内病的バリアント
母性継承性のCDKN1Cの病的バリアント、父性継承性のIGF2の病的バリアント、両親いずれからの継承も考えられるPLAG1やHMGA2の病的バリアントについては、絨毛採取もしくは羊膜穿刺で得たサンプルを用いて着床前遺伝学的検査や出生前遺伝学的検査を行うことで診断が可能である[Eggermannら2016,Abi Habibら2018]。
注:着床前遺伝学的診断の結果や、SRS関連の遺伝学的変化の出生前検査の結果は、出生後の臨床所見を予測するものとして信頼性をもって使用することはできない[Eggermannら2016]。SRS患児の成長ホルモン感受性はさまざまであり、遅発性の追いつき成長の量にも幅があり、発達障害の程度もさまざまである。
出生前検査の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。
早期診断を目的とするのではなく、堕胎を目的としてこれを利用しようという場合は、特にそれが言える。現在、多くの医療機関では、出生前検査を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- Genetic and Rare Diseases Information Center (GARD)
Phone: 301-251-4925; 888-205-2311
Silver-Russell syndrome
- L'AISRS, Associazione Italiana Sindrome di Russell-Silve
rItaly
www.aisrs.it
- L'ASBL ALICE: Association Libre d'Informations sur la Croissance des Enfants Silver Russell
Belgium
www.alice.be
- L'Association Française des Familles touchées par le syndrome de Silver Russell (SSR)
France
www.silver-russell.fr
- National Organization for Rare Disorders (NORD)
Russell Silver Syndrome
Silver Russell Syndrome Global Alliance
www.silverrussellsyndrome.org
- Bundesverband Kleinwüchsige Menschen (BKMF)
Leinestrasse 2
28199 Bremen
Germany
Phone: 49-421-336169-0
Fax: 49-421-336169-18
Email: info@bkmf.de
www.bkmf.de
- Child Growth Foundation
United Kingdom
Phone: 0208 995 0257
Email: nfo@childgrowthfoundation.org
www.childgrowthfoundation.org
- Human Growth Foundation (HGF)
997 Glen Cove Avenue
Suite 5
Glen Head NY 11545
Phone: 800-451-6434 (toll-free)
Fax: 516-671-4055
Email: hgf1@hgfound.org
www.hgfound.org
- Little People of America, Inc. (LPA)
250 El Camino Real
Suite 201
Tustin CA 92780
Phone: 888-572-2001 (toll-free); 714-368-3689
Fax: 714-368-3367
Email: info@lpaonline.org
www.lpaonline.org
- MAGIC Foundation
4200 Cantera Drive #106
Warrenville IL 60555
Phone: 800-362-4423 (Toll-free Parent Help Line); 630-836-8200
Fax: 630-836-8181
Email: contactus@magicfoundation.org
Russell Silver Syndrome
- Silver-Russell Support Group
c/o Child Growth Foundation
2 Mayfield Avenue
Chiswick WA 1PW
United Kingdom
Phone: 020 8995 0257; 020 8994 7625
Fax: 020 8995 9075
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:Silver-Russell症候群の遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | 座位別データベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| CDKN1C | 11p15.4 | サイクリン依存性キナーゼ阻害因子 | CDKN1C database | CDKN1C | CDKN1C |
| H19 | 11p15.4 | 不明 | H19@LOVD | H19 | H19 |
| HMGA2 | 12q14.3 | 高移動度群タンパク質HMGI-C | HMGA2 database | HMGA2 | HMGA2 |
| IGF2 | 11p15.5 | インスリン様成長因子Ⅱ | LOVD-Growth Consortium(IGF2) | IGF2 | IGF2 |
| PLAG1 | 8q12.1 | ジンクフィンガータンパク質PLAG1 | PLAG1 database | PLAG1 | PLAG1 |
| 不明 | 7番染色体 | 不明 |
データは、以下の標準資料から作成したものである。遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:Silver-Russell症候群関連のOMIMエントリー(内容の閲覧はOMIMへ)
| 103280 | H19, IMPRINTED MATERNALLY EXPRESSED NONCODING TRANSCRIPT; H19 |
| 147470 | INSULIN-LIKE GROWTH FACTOR II; IGF2 |
| 180860 | SILVER-RUSSELL SYNDROME 1; SRS1 |
| 600698 | HIGH MOBILITY GROUP AT-HOOK 2; HMGA2 |
| 600856 | CYCLIN-DEPENDENT KINASE INHIBITOR 1C; CDKN1C |
| 603026 | PLAG1 ZINC FINGER PROTEIN; PLAG1 |
| 618905 | SILVER-RUSSELL SYNDROME 2; SRS2 |
| 618907 | SILVER-RUSSELL SYNDROME 4; SRS4 |
| 618908 | SILVER-RUSSELL SYNDROME 5; SRS5 |
分子レベルの病原
染色体11p15.5関連のSilver-Russell症候群(SRS)
胎児の成長に関して、染色体11p15.5のインプリンティング遺伝子が重要であることが知られている[DeChiaraら1990,Fitzpatrickら2002,Eggermannら2009]。
インプリンティングを受けた遺伝子はしばしばクラスターで存在し、その中にインプリンティング制御領域(ICR)が存在する。11p15.5のインプリンティング遺伝子のクラスターの1つにおいては、インプリンティング制御領域1(ICR1)の片親特異性の可変メチル化により、IGF2とH19の相補的発現制御が行われている。なお、IGF2は胎児の成長に決定的に重要な成長因子の1つをコードしており、H19は非コード転写産物のコード配列である。SRSでは、ICR1の低メチル化によりH19の両アレル発現とIGF2のサイレンシングが起こり、その結果、成長抑制が生じる。このタイプはSRS罹患者の30%-60%を占める[Gincquelら2005,Abu-Ameroら2010,Wakelingら2017]。
11p15.5関連のSilver-Russell症候群でみられる分子レベルの変化については、図2に図式を示したので参照されたい。
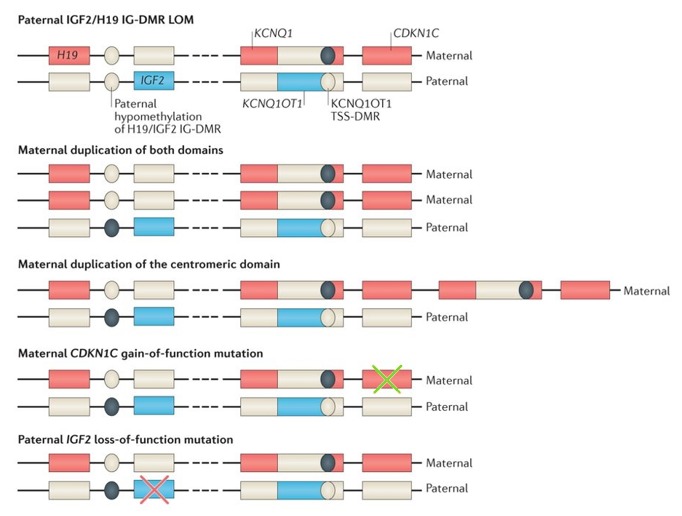
図2:
H19/IGF2遺伝子間メチル化可変領域に父性の低メチル化が起こることにより、父親由来のIGF2の発現が停止し、本来、母性発現であるはずのH19の発現過剰が生じる。これにより、成長抑制という表現型が形成されるに至る[Gicquelら2005,Wakelingら2017]。稀ながら、母性のセントロメア側の重複ないし2つのドメインの重複により、CDKN1Cの発現過剰が生じるタイプのものもある。こちらも稀ながら、家族性の例の中には、母性のCDKN1Cの機能獲得型病的バリアント(緑の×印)[Brioudeら2013,Wakelingら2017]、あるいは父性のIGF2の機能喪失型病的バリアント(赤の×印)[Begemannら2015,Wakelingら2017]によって生じるタイプのものもある。
この図は、Wakelingら[2017]の論文から引用したものである(説明文は割愛している)。この図は、クリエイティブコモンズ【表示4.0国際】ライセンスのもとに公開されている。
11p15.5のメチル化異常にはいくつかのメカニズムが存在する。
- 父親由来染色体のICR1の低メチル化がSRS罹患者の30%-60%にみられる。ICR1は、IGF2、H19両方のメチル化を制御しているので、可変領域分析を行うと、大多数の症例で両遺伝子に低メチル化がみられる。
注:(1)父親のICR1に生じる11p15.5の低メチル化は、通常、配偶子形成後に生じるものなので、大多数のSRS罹患者については、メチル化異常が体細胞全体に及ぶ分布パターンとなる(検査の指針については表1を参照)。11p15.5 ICR1の低メチル化を示す罹患者の約1%は、ICR1領域の部分欠失を有している。こうした稀な例で、エピゲノムの異常の背景に遺伝子の問題の存在が同定されることがある[Abi Habibら2017]。
(2)少数のSRS罹患者では、H19、IGF2にそれぞれ限局した選択的低メチル化を呈する。
- SRS罹患者のごく少数で、11p15.5領域の母性の重複を有している例がみられる。転座や逆位を伴うようなもっと大きな重複であれば、細胞遺伝学的分析で探知可能である[Fisherら2002,Eggermannら2005]。また、微小欠失・微小重複についてはSNPマイクロアレイで探知できる[Begemannら2012,Eggermannら2012](表1)。
- 稀な家族性の例で、背景に次のようなメカニズムの存在が報告されている。
- 母性遺伝の11p15の重複
- 母性遺伝のCDKN1Cの機能獲得型バリアント
- 父性遺伝のIGF2の機能喪失型バリアント
- 11p15.5 ICR1の微小欠失
こうした家系では、再発リスクは最大50%となりうる。したがって、11p15.5脱メチル化を有する罹患者においては、背景にあるコピー数多型の探索が重要となる[Abi Habibら2017,Wakelingら2017,Heideら2018]。
7番染色体関連のRussel-Silver症候群
母性UPD7で疾患原因となる遺伝子座として、少なくともPEG1/MESTは含まれているように思われる。PEG1/MESTは、7q32に座位するインプリンティングを受けた遺伝子クラスターである[Hannulaら2001,Eggermannら2012]。7q32のメチル化異常を生じるメカニズムとしては、いくつかの可能性がある。
- 母性UPD7
母性UPD7がSRSの7%-10%で報告されている[Mooreら1999,Hannulaら2001,Kimら2005]。その成り立ちには次のものがある。
- 母性のアイソダイソミーもしくはヘテロダイソミー[Bernardら1999,Priceら1999]
- upd(7)matのモザイク[Reboulら2006]
- 分節型upd(7)matの報告もみられる。
- 7q31-qterの母性UPD7が1症例[Hannulaら2001]
- 7番染色体長腕の大部分である7q11.2-qterの母性UPDが2症例[Eggermann 2008]
- 稀な7番染色体の異常
SRS罹患者でみられる稀な7番染色体の異常としては次のようなものがある。
- 母性ヘテロダイソミーを伴う7番染色体トリソミーモザイクの子どもが2症例[Floriら2005,Font-Montgomeryら2005]。このうちの1例は出生前にこれが同定されている[Font-Montgomeryら2005]。
- 7番染色体長腕の中間部欠失である del(7)(q21.1q21.3) の子どもが1例
- 蛍光in situハイブリダイゼーション法で同定された、7p11.2-p12の顕微鏡レベル以下の(通常の核型分析では見えない)重複[Joyceら1999,Monkら2000]
更新履歴:
-
Gene Reviews著者: Howard M Saal, MD, Madeleine D Harbison, MD, and Irene Netchine, MD, PhD.
日本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2019.10.21. 日本語訳最終更新日: 2024.4.14.[in present]
![]()