








Diagnostic Imaging of White Matter Abnormalities
Junichi TAKANASHI, Department of Pediatrics, Tokyo Women's Medical University, Yachiyo Medical Center
Introduction
In this paper, I outline the approach employed for diagnosis of disorders that appear as abnormal signals in the cerebral white matter on magnetic resonance imaging (MRI), from imaging to diagnosis. Disorders that mainly affect the white matter are generally referred to as "leukoencephalopathy" or "white matter disorders" in English.1, 2) 3) Another term, leukodystrophy, is sometimes confused with white matter degeneration, but this actually refers to a narrower spectrum of disorders with a genetic component (inherited demyelinating disorders).
Imaging-based classification of white matter disorders
The advent of MRI has dramatically improved our ability to detect lesions in the white matter of the central nervous system. Many known forms of white matter disorders exhibit specific signs on MRI, which is useful for their diagnosis. Identifying patterns of white matter abnormality seen on MRI (T1-weighted, T2-weighted, or FLAIR imaging) makes it easy to narrow down the possibilities in multiple differential diagnoses. Schiffmann and van der Kamp's classification of white matter disorders according to MRI findings is of practical value2, 4) (Figure 1, Table 1). Even if this does not lead to a final diagnosis, the classification of imaging findings may lead to the later discovery of a new disorder. Here, I describe white matter disorders in terms of the above MRI classifications, and explain the main types of disorders.
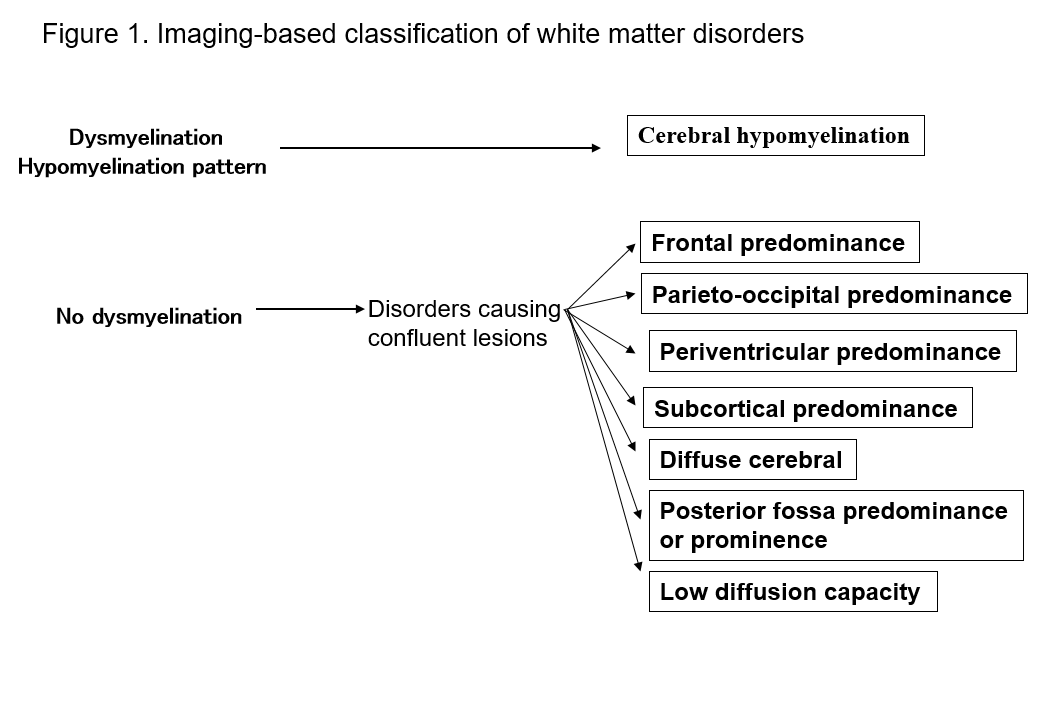
Table 1. List of disorders by MRI patterns
- Frontal predominance
Alexander disease, frontal variant of X-linked adrenoleukodystrophy (ALD), metachromatic leukodystrophy (MLD), neuroaxonal leukodystrophy with spheroids. - Parieto-occipital predominance
X-linked adrenoleukodystrophy (ALD), Krabbe disease,early onset peroxisomal disorders, neonatal hypoglycemia. - Periventricular predominance
Metachromatic leukodystrophy (MLD), Krabbe disease, Sjögren-Larsson syndrome, adult polyglucosan body disease, leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL), periventricular leukomalacia (PVL), HIV encephalopathy, later-onset neuronal ceroid lipofuscinoses. - Subcortical predominance
L-2-hydroxyglutaric aciduria, galactosemia, Kearns-Sayer syndrome, propionic academia, urea cycle disorders, Canavan disease. - Diffuse cerebral
Megalencephalic leukoencephalopathy with subcortical cysts (MLC), leukoencephalopathy with vanishing white matter (VWM), merosin deficient congenital muscular dystrophy, mitochondrial disease, molybdenum cofactor deficiency, sulfite oxidase deficiency, advanced cases of white matter disorders. - Posterior fossa predominance or prominence
Lesions of the cerebellum and cerebellar peduncles: cerebrotendinous xanthomatosis (CTX), peroxisomal disorders, Alexander disease, leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL), maple syrup urine disease, histiocytosis, adult autosomal dominant leukodystrophy related to a lamin B1 duplication, heroin and cocaine toxicity.
Brainstem lesions: Alexander disease, LSBL, peroxisomal disorders, Wilson disease, adult polyglucosan disease, Leigh syndrome, dentatorubropallidoluysian atrophy (DRPLA), adult polyglucosan body disease, adult autosomal dominant leukodystrophy related to a lamin B1 duplication. - Multifocal lesions
TORCH syndrome (congenital cytomegalovirus infection), brucellosis, acute disseminated encephalomyelitis (ADEM), multiple sclerosis (MS), neuromyelitis optica (NMO), cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), atherosclerosis, amyloid angiopathy, COL4A1-associated cerebral small-vessel disease, Fabry disease, Susac syndrome, mitochondrial disease, L-2-hydroxyglutaric aciduria, mucopolysaccharidosis (MPS), chromosomal abnormalities (such as 6p-syndrome). - Lesions with low diffusion capacity
Maple syrup urine disease, methionine adenosyltransferase I/III deficiency, phenylketonuria, non-ketotic hyperglycinemia, Canavan disease, active lesions in Krabbe disease, and metachromatic leukodystrophy.
1. Cerebral white matter hypomyelination
This refers to a group of disorders in which the formation of the myelin sheath is impaired or delayed, and its images resemble those of newborn babies with immature myelination. On T2-weighted images, the white matter characteristically appears as a widespread hyperintensity that is faint compared with the cortex. For more details, see the website of congenital cerebral hypomyelinatio.
If white matter lesions are not consistent with cerebral white matter hypomyelination, it must be determined whether they are confluent or multiple.2) Confluent white matter lesions are usually due to inherited white matter degeneration (leukodystrophy) and in most cases are bilaterally symmetrical. Multiple white matter lesions are usually asymmetrical and acquired. Confluent white matter lesions are further subdivided into categories 2–7 below.
2. Frontal predominance
In this group of disorders, extensive white matter lesions are present predominantly in the frontal lobe. They include Alexander disease, the frontal variant of X-linked adrenoleukodystrophy (ALD), metachromatic leukodystrophy (MLD), and neuroaxonal leukodystrophy with spheroids.
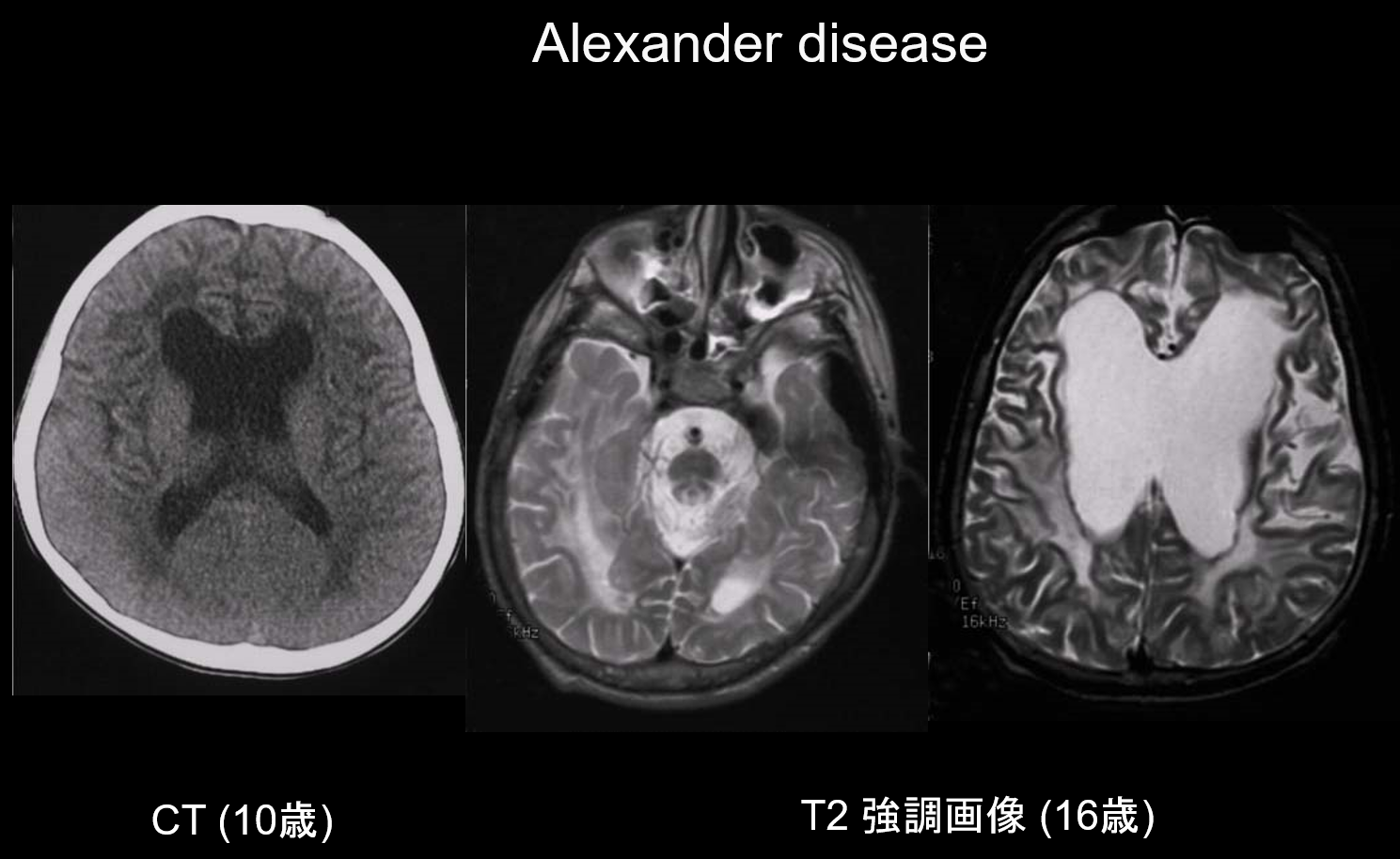
(a) Alexander disease.
Alexander disease is an autosomal dominant inherited disorder caused by a mutation in the GFAP gene on chromosome 17q21. It results in the accumulation of Rosenthal fibers in the stellate glial cells. These fibers are composed of GFAP and stress proteins (αB-crystallin and HSP27. Alexander disease mainly occurs in infancy, between the ages of 3 months and 2 years, with the appearance of megalencephaly, developmental retardation, spastic palsy, and epilepsy. On MRI, it may exhibit (i) widespread white matter lesions, predominantly in the frontal lobe; (ii) T1 hyperintense and T2 hypointense margination around the lateral ventricles; (iii) lesions in the basal ganglia and thalamus; (iv) brainstem lesions; and (v) contrast enhancement of active lesions (Figure 2). In the early stages, along with white matter and putamen lesions, swelling is seen which may gradually cause atrophy or cyst formation.
3. Parieto-occipital predominance
The main characteristic of this group of disorders is parieto-occipital white matter lesions. They include X-linked adrenoleukodystrophy (ALD), Krabbe disease, early onset peroxisomal disorders, and neonatal hypoglycemia.
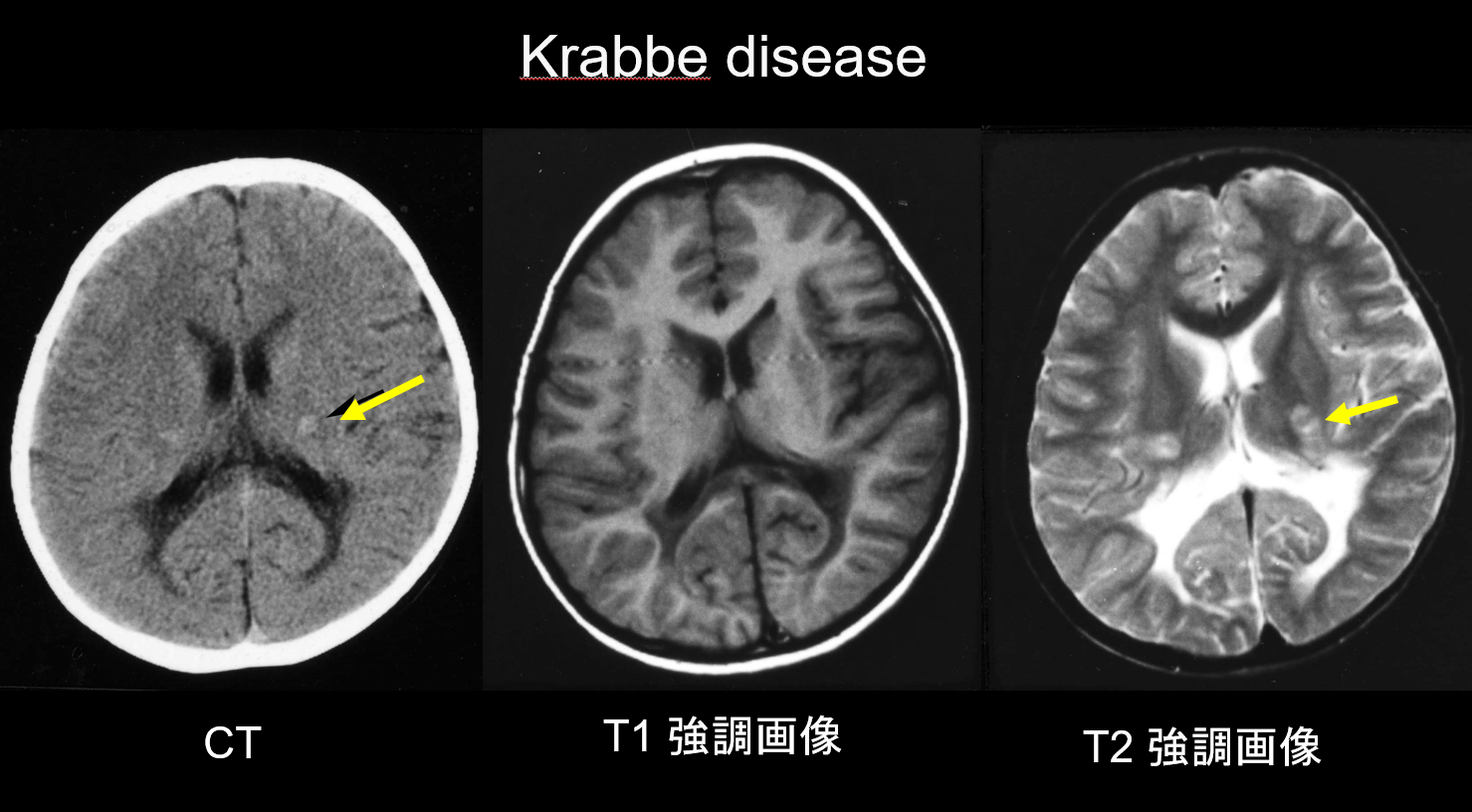
(a) Krabbe disease.
Krabbe disease is an autosomal recessive inherited disorder (lysosomal storage disease) caused by galactosylceramidase deficiency (chromosome 14q31), in which the accumulation of highly cytotoxic psychosine is believed to cause widespread demyelination. Large, multinucleated cells called "globoid cells" also appear. Depending on the age at which it appears, it is classified as infantile, late-onset infantile, juvenile-onset, or adult-onset disease. Most cases are infantile and start with the appearance of fever, irritability, difficulty feeding, developmental retardation, peripheral neuropathy, spasticity, and optic nerve atrophy at age 3–6 months. During the early stages, computed tomography (CT) reveals characteristic hyperdensity in the thalamus and corona radiata. This is believed to reflect high-density globoid cells and glial proliferation. MRI may also show T1 hyperintensity and T2 hypointensity around the ventricles, as well as linear structures similar to those seen in MLD (Figure 3). The cerebellar dentate nucleus, cerebellar white matter, and brainstem pyramidal tract exhibit T2 hyperintensity from an early stage.
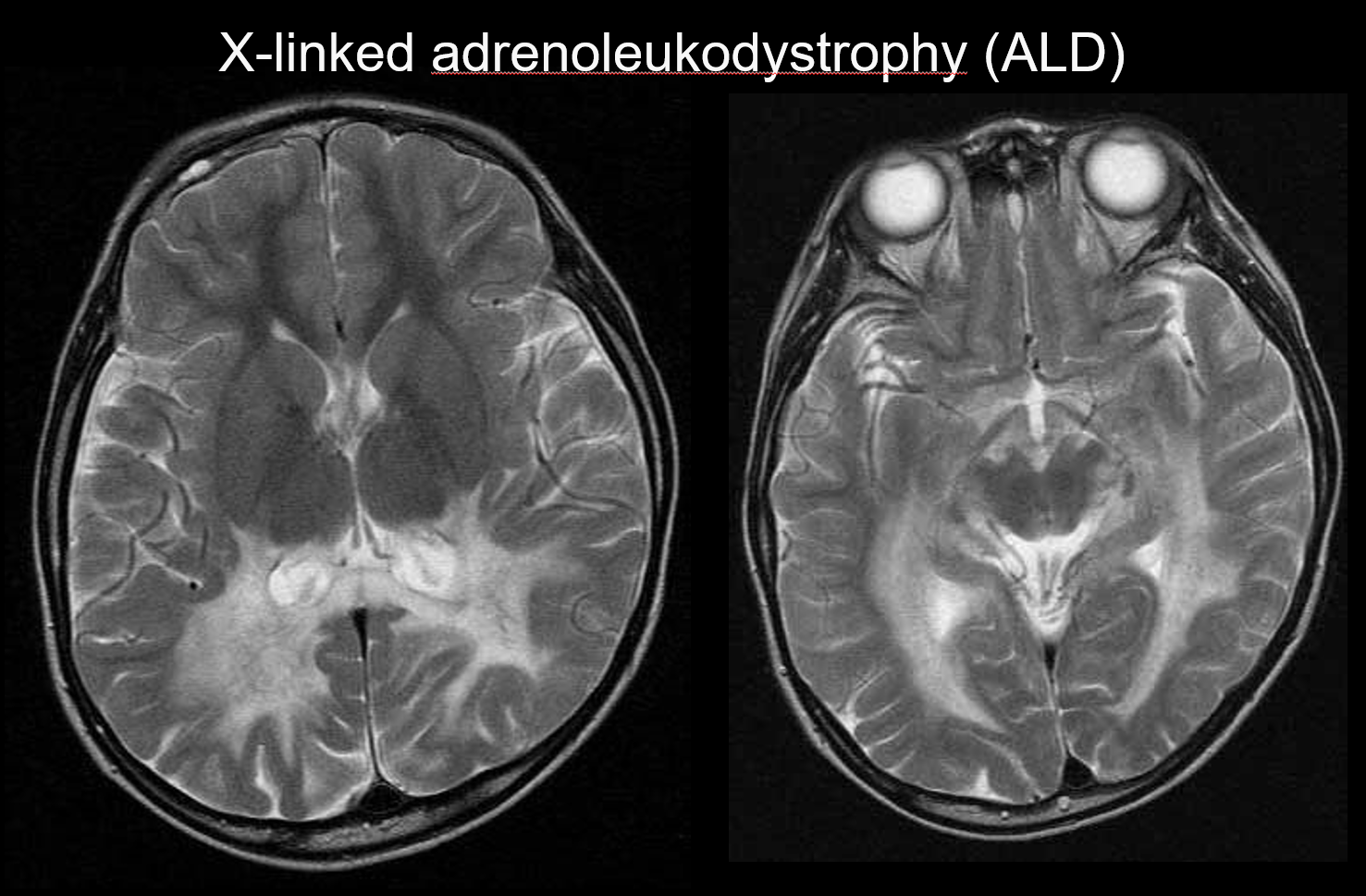
(b) X-linked adrenoleukodystrophy
X-linked adrenoleukodystrophy (ALD) is an X-linked recessive inherited disorder (peroxisomal disorder) caused by an abnormality of the ABCD1 gene (chromosome Xq28). Impaired β-oxidation results in the accumulation of very-long-chain fatty acids in the cerebral white matter and the adrenal glands, causing demyelination and adrenal insufficiency. ALD is categorized into childhood, adolescent, and adult cerebral forms, adrenomyeloneuropathy (AMN), and Addison disease only. The childhood cerebral form develops at 5–8 years of age, with the appearance of symptoms including intellectual deterioration, spastic gait, and impaired vision and hearing. Pathologically, demyelination progresses from the white matter surrounding the trigone of the lateral ventricle to the splenium of the corpus callosum, gradually extending anterolaterally. Reflecting the disease pathology, symmetrical T2 hyperintensities and T1 hypointensities extending anterolaterally from the white matter surrounding the trigone of the lateral ventricle are apparent on MRI, with contrast enhancement evident at the margins (Figure 4). Corticospinal tract lesions are also evident.
4. Periventricular predominance
These disorders are mainly characterized by lesions in the white matter surrounding the lateral ventricles, with the subcortical white matter (U-fibers) being preserved. This pattern is seen in numerous disorders, including MLD, and is therefore comparatively non-specific. Mildly abnormal signals around the lateral ventricles are also seen in cortical degeneration, particularly neuronal ceroid lipofuscinoses that develop post-infancy.
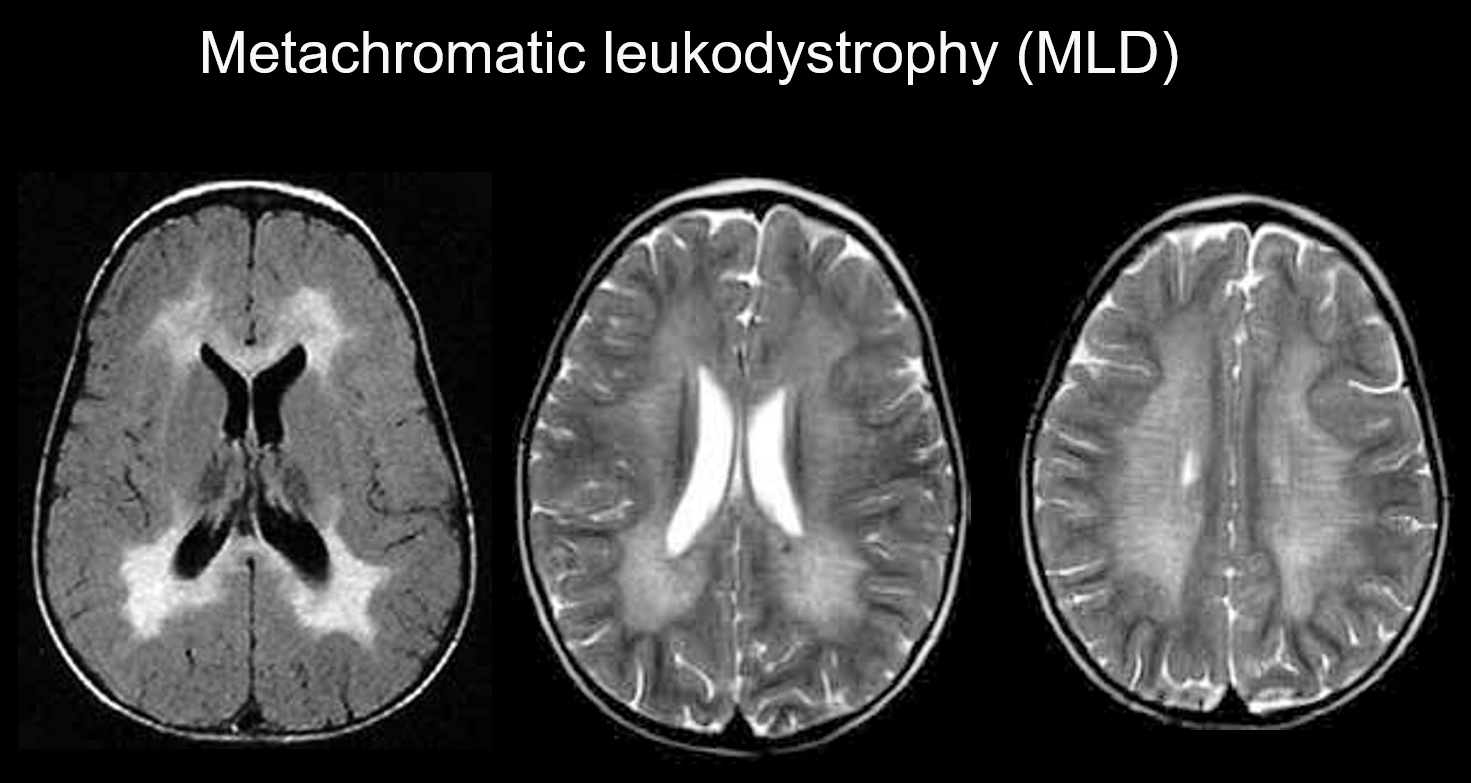
(a) Metachromatic leukodystrophy.
Metachromatic leukodystrophy is an autosomal recessive inherited disorder (lysosomal storage disorder) caused by arylsulfatase-A deficiency (chromosome 22q13.31), in which the accumulation of highly toxic sulfatide results in demyelination. Depending on the age at which it appears, it is classified as congenital, infantile-onset, juvenile-onset, or adult-onset. Its symptoms include cognitive regression, spastic palsy, involuntary movements, peripheral neuropathy, and optic nerve atrophy. It appears on T2-weighted imaging as hyperintensities of the white matter mainly around the lateral ventricles, and on T1-weighted imaging as mild hypointensities. The lesions tend to be predominantly in the frontal lobe. Bands of normal intensity (tiger stripes) may be evident within the widespread abnormal signals in the white matter (Figure 5). These are believed to be due to the partial preservation of the myelin sheath in the perivascular space and to the accumulation of myelin sheath breakdown products in macrophages.
5. Subcortical predominance
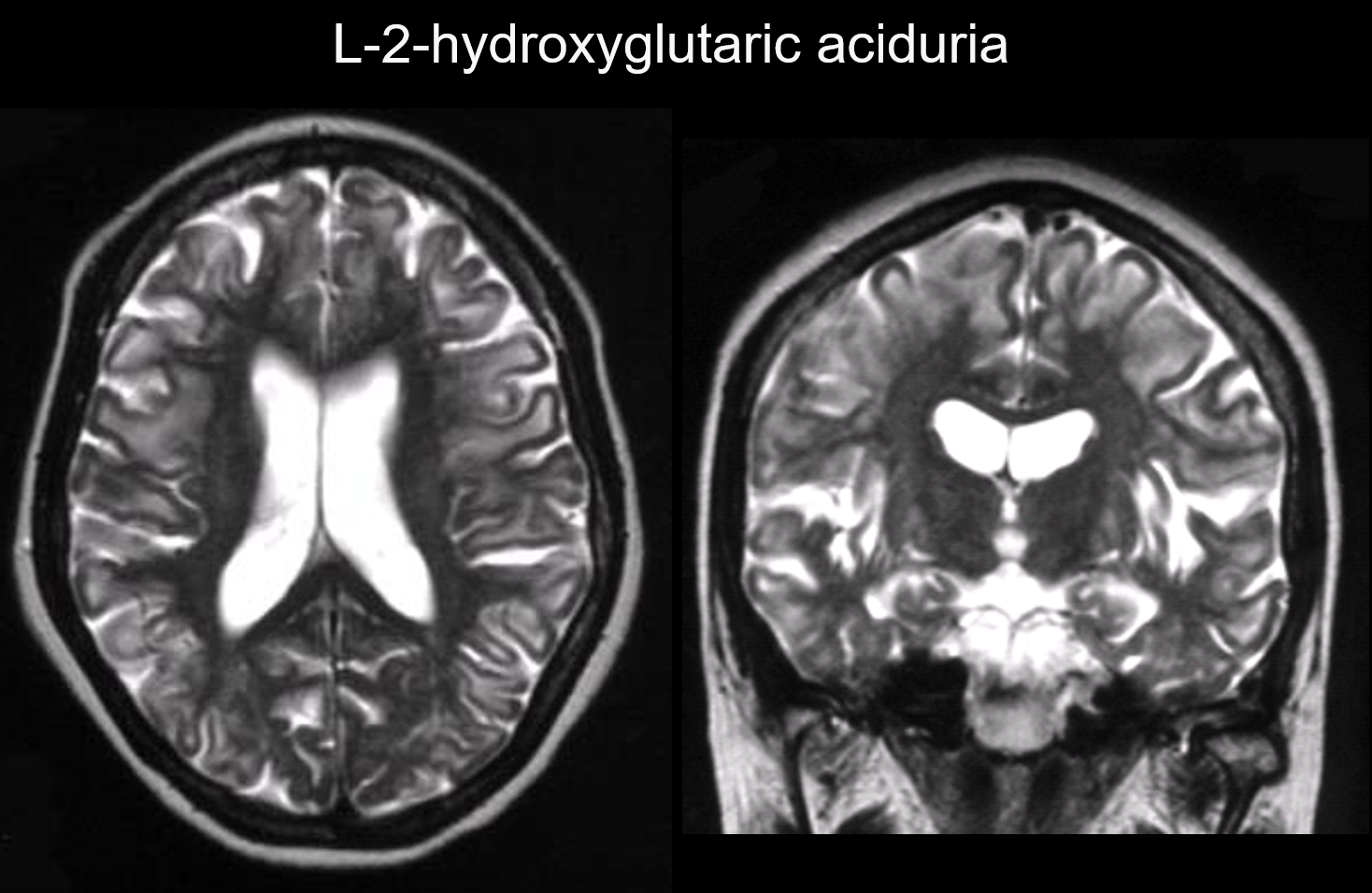
In these disorders, lesions occur primarily in the subcortical white matter, including the U-fibers. Disorders with this pattern include L-2-hydroxyglutaric aciduria (Figure 6), galactosemia, Kearns-Sayer syndrome, propionic academia, urea cycle disorders, and early-stage Canavan disease.
6. Diffuse cerebral
In these disorders, abnormal signals appear throughout the cerebral white matter. They exhibit strong T2 hyperintensities compared with the T2 signals produced by unmyelinated white matter (hypomyelination). In addition to cases of megalencephalic leukoencephalopathy with subcortical cysts and leukoencephalopathy with vanishing white matter, patients with any type of white matter disorder eventually exhibit this pattern as the disease advances.
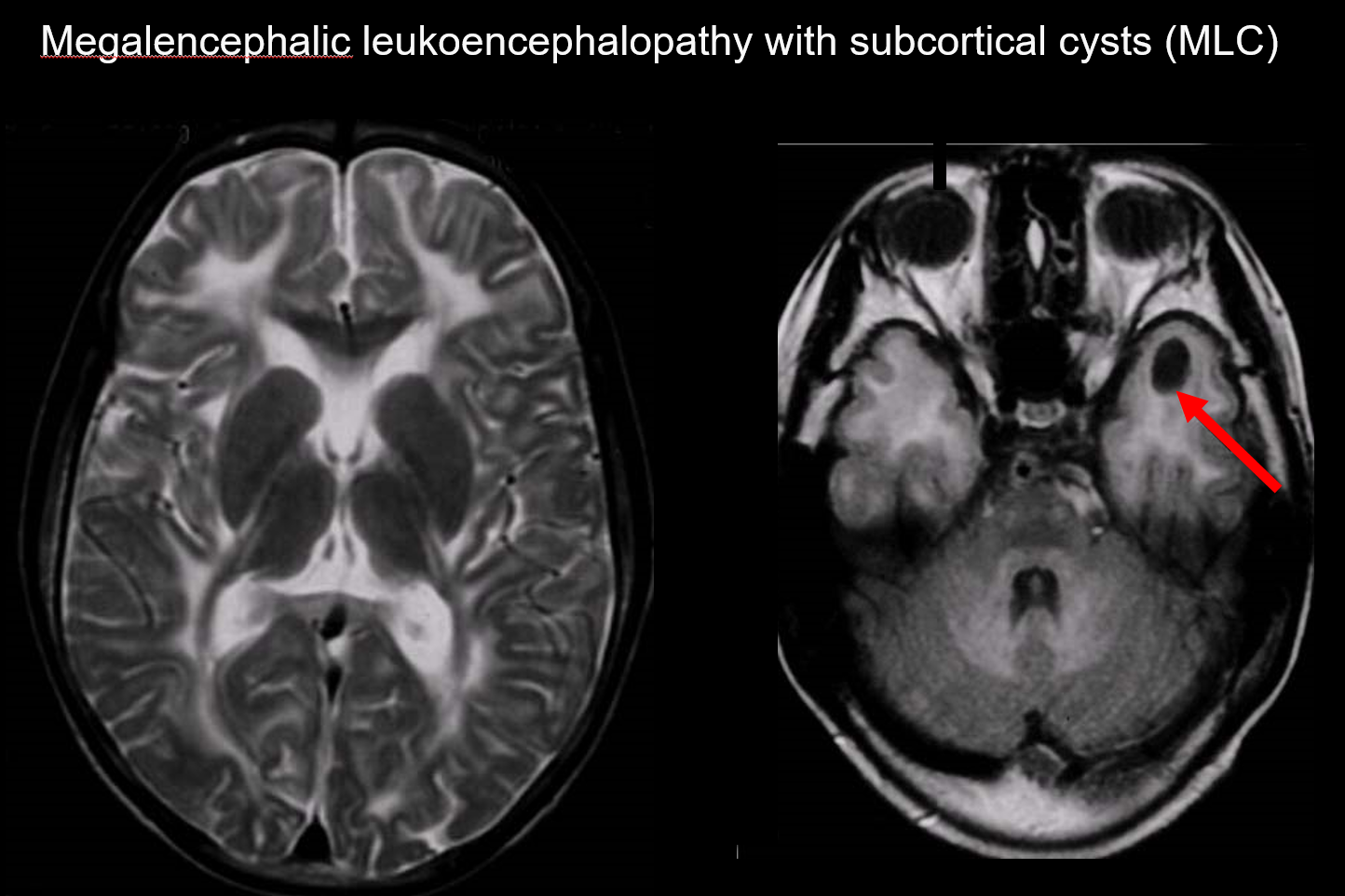
(a) Megalencephalic leukoencephalopathy with subcortical cysts (MLC)
MLC is an autosomal recessive inherited disorder caused by an abnormality of the MLC1 gene, and its onset in infancy is marked by megalocephaly, slowly progressing motor deterioration, ataxia, and spasticity. MRI reveals characteristic widespread abnormal signals in the white matter and mild swelling of the white matter, as well as cyst formation in the parietal and temporal lobes (Figure 7).7, 8) T1-weighted and T2-weighted imaging reveal abnormal white matter, while the cysts all exhibit T1 hypointensity and T2 hyperintensity, making them particularly difficult to detect. FLAIR imaging, which visualizes cysts (water) as hypointensities, is valuable for its diagnosis. It is more common among Japanese than vacuolating megalencephalic leukoencephalopathy.
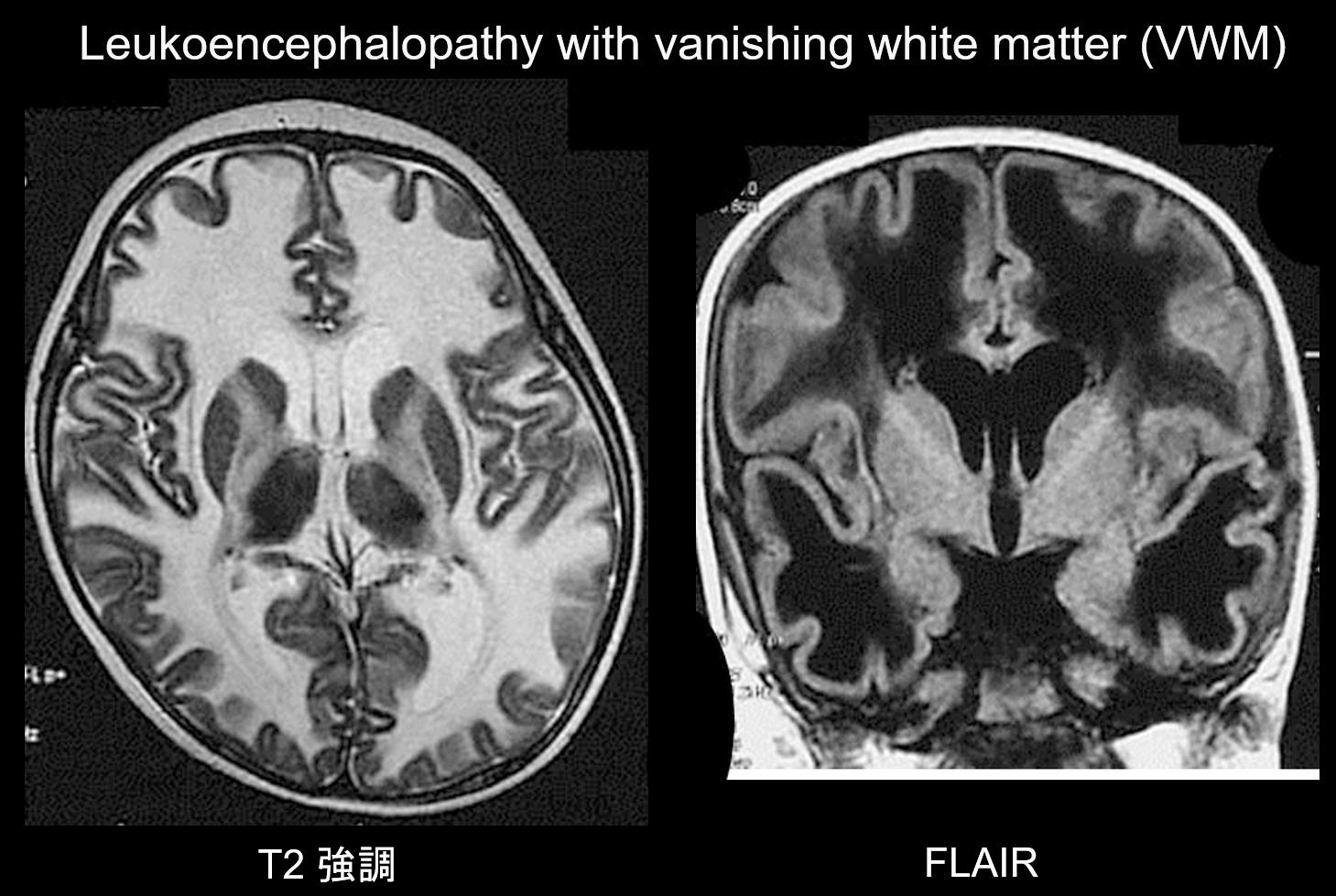
(b) Leukoencephalopathy with vanishing white matter.
Leukoencephalopathy with vanishing white matter (VWM) is an autosomal recessive inherited disorder caused by a deficiency of eIF2B, a protein associated with eIF2, which transfers initiator tRNA to ribosomes. eIF2B consists of five different proteins, which all have different genetic loci. VWM has been shown to be the same disorder as childhood cerebellar ataxia and central hypomyelination (CACH). Patients are normal during the neonatal period and early infancy, but post-onset (usually at age 2–6 years) they develop slowly progressive cognitive regression, spasticity, and ataxia. These symptoms are known to be exacerbated by infection or minor trauma. The cerebral white matter exhibits widespread T2 hyperintensity and T1 hypointensity and is gradually replaced by fluid over time (as the name implies, the white matter vanishes) (Figure 8). The cystic white matter contains banded structures that are believed to represent the remaining tissue. Abnormal signals are also seen in the brainstem, particularly the central tegmental tract. FLAIR imaging is valuable for the diagnosis of this disorder.
7. Posterior fossa predominance or prominence
These disorders are characterized by lesions predominantly in the brainstem and the cerebellum. Cerebellar white matter lesions may be caused by disorders including cerebrotendinous xanthomatosis (CTX), peroxisomal disorders, Alexander disease, leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL), maple syrup urine disease, histiocytosis, and heroin and cocaine toxicity. Brainstem lesions may be caused by disorders such as Alexander disease, LSBL, and adult polyglucosan disease. Middle cerebellar peduncle lesions are seen in fragile X syndrome and adult autosomal dominant leukodystrophy related to a lamin B1 duplication.
8. Multifocal lesions
Unlike disorders that produce the confluent lesions described in 2–7 above, the disorders in this section result in multifocal (scattered) lesions. They include infections such as TORCH syndrome (due to congenital cytomegalovirus infection or other cause) and brucellosis; inflammatory disorders such as acute disseminated encephalomyelitis (ADEM), multiple sclerosis (MS), and neuromyelitis optica (NMO); vasculopathies such as cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), atherosclerosis, amyloid angiopathy, COL4A1-associated cerebral small-vessel disease, Fabry disease, and Susac syndrome; and inherited conditions such as mitochondrial disease, L-2-hydroxyglutaric aciduria, mucopolysaccharidosis (MPS), and chromosomal abnormalities (such as 6p-syndrome).
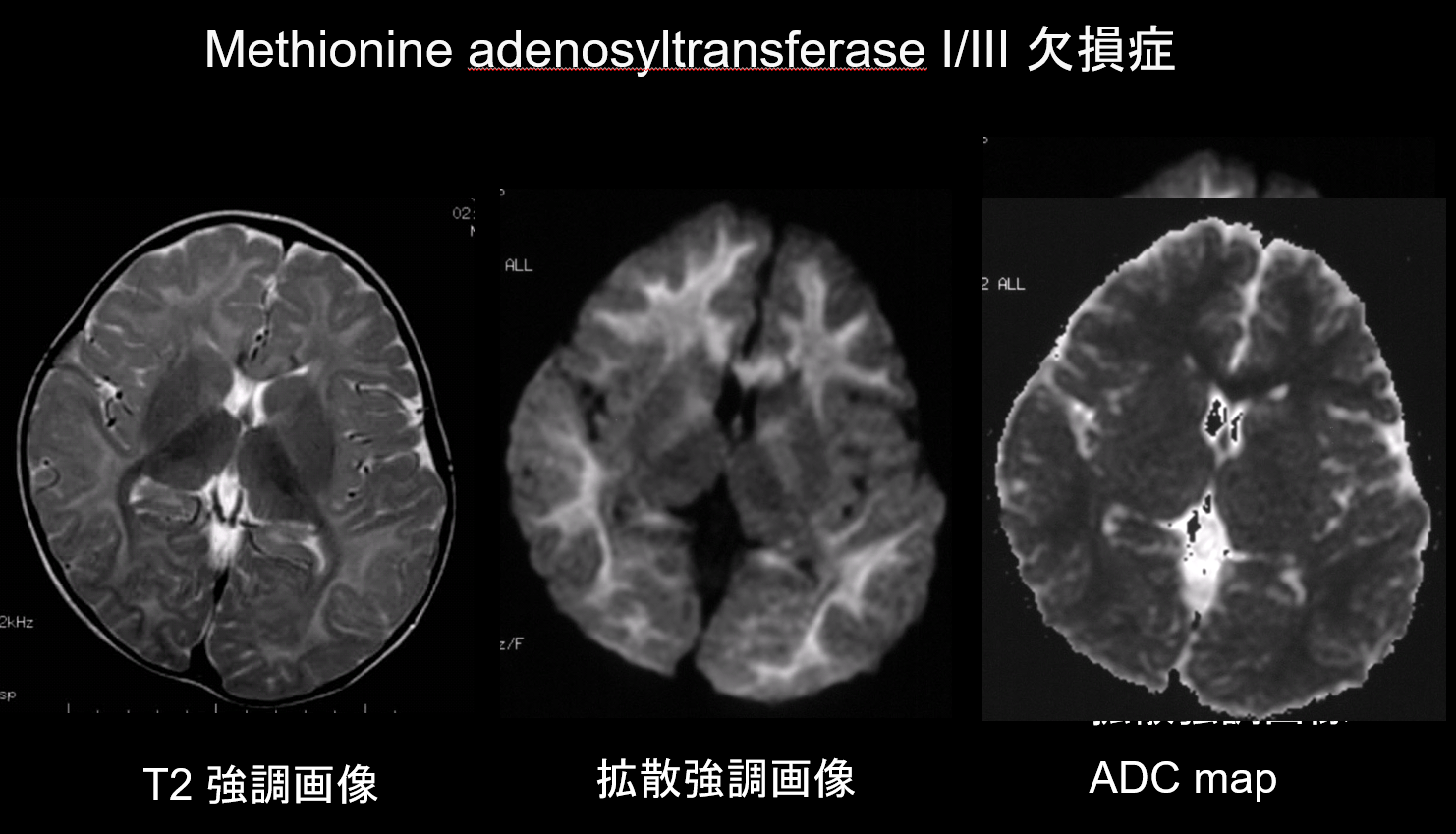
9. Lesions with low diffusion capacity
In both demyelination and hypomyelination, the main pathologies of white matter disorders, there is a decrease in the amount of myelin, which limits diffusion, and the corresponding increase in extracellular fluid results in T2 hyperintensities with a high apparent diffusion coefficient (ADC). It is rare for white matter disorders to exhibit both T2 hyperintensities and low ADC, and this combination is therefore of high diagnostic value. Disorders characterized by the presence of intramyelinic edema within the myelin sheath and in the gaps between sheaths display a low ADC. They include maple syrup urine disease, methionine adenosyltransferase I/III deficiency (Figure 9), phenylketonuria, non-ketotic hyperglycinemia, and Canavan disease. Krabbe disease and metachromatic leukodystrophy may also exhibit low ADC in some white matter lesions, as intramyelinic edema may occur during the acute phase of demyelination.
References
- Van der Knaap MS, Valk J. Classification of myelin disorders. In Van der Knaap MS, Valk J, eds. Magnetic resonance of myelination and myelin disorders. 3rd ed. Berlin: Springer, 2005, 20-24.
- Schiffmann R, van der Knaap MS. An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009; 72: 750-759
- Takanashi J. Diagnostic imaging of white matter disorders. Journal of the Japan Pediatric Society 2007; 111: 1243-1254. [In Japanese]
- Van der Knaap MS, Breiter SN, Naidu S, et al. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology 1999; 213: 121-133.