


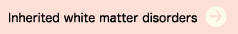






Clinical diagnosis of patients with cerebral white matter abnormalities
Masayuki SASAKI, MD
Department of Child Neurology, National Center of Neurology and Psychiatry
1. Summary and Definitions
Primary cerebral white matter abnormalities mainly consist of disorders that damage the myelin sheath in the central nervous system. Many are inherited degenerative or metabolic disorders. They include cerebral white matter degeneration, demyelination, and hypomyelination.
2. Etiology
Although the causative gene has been identified in some cases, in others it remains unknown. Classically, these disorders are categorized in terms of the proteins that form the myelin sheath of the central nervous system (proteolipid protein [PLP] and myelin basic protein [MBP]), subcellular organelles (e.g., peroxisomes, lysosomes, mitochondria), and metabolic substances (abnormalities of amino or organic acid metabolism), and abnormalities of genes that code for various functional proteins within the cell (Table 1).
Table 1. Classic classification of primary cerebral white matter disorders with representative diseases
- Myelin dysproteinemias
Pelizaeus-Merzbacher disease (proteolipid protein, PLP)
18q- deletion syndrome (myelin basic protein, MBP) - Lysosomal storage diseases
Krabbe disease (globoid cell leukodystrophy, GLD)
Metachromatic leukodystrophy (MLD)
GM1/GM2 gangliosidosis - Peroxysomal diseases
X-linked adrenoleukodystrophy (ALD)
Zellweger syndrome - Mitochondrial disease
Kearns-Sayre syndrome
Progressive cavitating leukoencephalopathy - Amino acid metabolism disorders
Phenylketonuria
Urea cycle disorders - Organic acid metabolism disorders
Canavan disease
L-2-hydroxyglutaric aciduria - Other metabolic disorders and degenerative diseases
Alexander disease
Hypomyelination with atrophy of basal ganglia and cerebellum (H-ABC)
Pol III-related leukodystrophies (4H syndrome, ADDH, TACH, LO, HCAHC)
Megalencephalic leukoencephalopathy with subcortical cysts (MLC)
Leukoencephalopathy with vanishing white matter (VWM)
Hypomyelination with congenital cataracts (HCC)
3. Clinical Symptoms
Age at onset varies depending on the type of disorder. Symptoms include delayed motor development or motor regression, spastic palsy, muscle hypertonia, and cognitive impairment (retardation or regression). Other symptoms such as seizures, cerebellar ataxia, involuntary movements (such as dystonia), and endocrine disturbances (short stature, delayed sexual maturation) may also be present.
4. Pathology
(a) Pathological classification
- "Demyelination" refers to the loss of a myelin sheath after its formation. Demyelination may occur in both the central and peripheral nervous systems.
- "Hypomyelination" refers to the presence of myelin sheaths that are not fully functional because of incomplete myelin formation. These sheaths easily break down at an early stage in the disorder. This term is used almost synonymously with "dysmyelination."
(b) MRI-based classification
Demyelination and hypomyelination can be distinguished to some extent by comparing the signal intensity of affected white matter on T1- and T2-weighted imaging, or by comparing its intensity with that of cortical white matter.1, 2)
- In demyelination, affected white matter appears strongly hyperintense on T2-weighted imaging and strongly hypointense on T1-weighted imaging. The cortical white matter (U-fibers) is usually preserved in the early stages of the disease.
- In hypomyelination, affected white matter is mildly hyperintense on T2-weighted imaging compared with the cerebral cortex, while on T1-weighted imaging it exhibits signal intensity varying from hyperintense (the normal pattern) to isointense and mildly hypointense. In most cases, the signal intensity is similar from the subcortical white matter to the deep periventricular white matter.
In addition to this basic imaging, cerebral white matter may increase or decrease in volume or become transparent, cystic, contrast-enhanced, or calcified, and these are all useful for differential diagnosis.
5. Diagnosis and Differential Diagnosis
The various disorders can be differentiated to some extent on the basis of age at onset, symptoms, and cranial MRI findings. The location of the white matter where abnormal signals are first evident (such as predominantly in the frontal lobe or the parietal lobe) is useful in reaching a diagnosis.
A. Demyelinating disorders
(a) Alexander disease
The main symptoms of the cerebral form of this disorder are an enlarged head circumference, seizures, and psychomotor retardation. It usually develops during the first year of life. As it progresses, spastic limb palsy and dysphagia also appear. Cranial imaging reveals abnormalities spreading out from the white matter in the frontal lobe (Figure 1). In many cases, the amount of white matter at the locations of the abnormal signals appears to be increasing. The brainstem form of this disease is most often seen in adults, and the abnormal signals in the cerebral white matter are not consistent in these cases.
Alexander disease is caused by a mutation in the GFAP gene, and demonstrates autosomal dominant inheritance. In the cerebral form, this is basically a de novo mutation. Since the advent of genetic diagnosis, confirmation of the presence of Rosenthal fibers by cerebral biopsy is no longer necessary.
(b) X-linked adrenoleukodystrophy
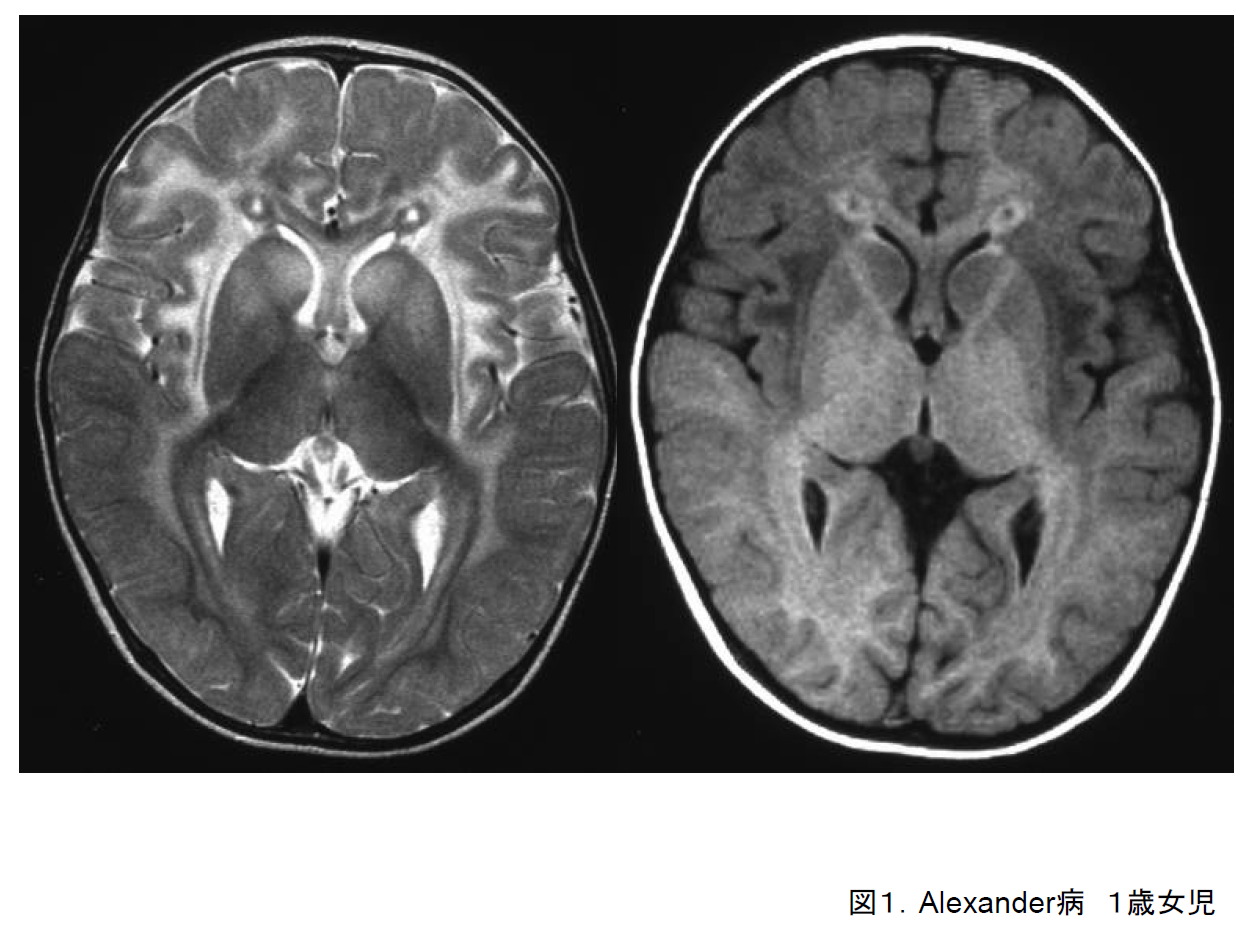
In the childhood cerebral form of X-linked adrenoleukodystrophy (ALD), symptoms including visual and motor impairment, declining academic performance, gait disorder, and seizures develop between the ages of 3 and 10. In most cases, the disease progresses over several years through quadriplegia to complete immobility. The adolescent cerebral form and the adult cerebral form are similar but develop at different ages.
There are various different variants of ALD, including the cerebral and spinal forms, and even members of the same family with the same mutation may develop a different form at a different age. On cranial MRI, demyelination is frequently seen to start from the deep white matter of the posterior and occipital lobes surrounding the posterior horn of the lateral ventricle (Figure 2-1), although on occasion it may start in the frontal lobe (Figure 2-2).
ALD is caused by an abnormality of the ABCD1 gene on chromosome Xq28 and demonstrates sex-linked recessive inheritance. Elevated levels of very-long-chain fatty acids in the blood are useful for its diagnosis.
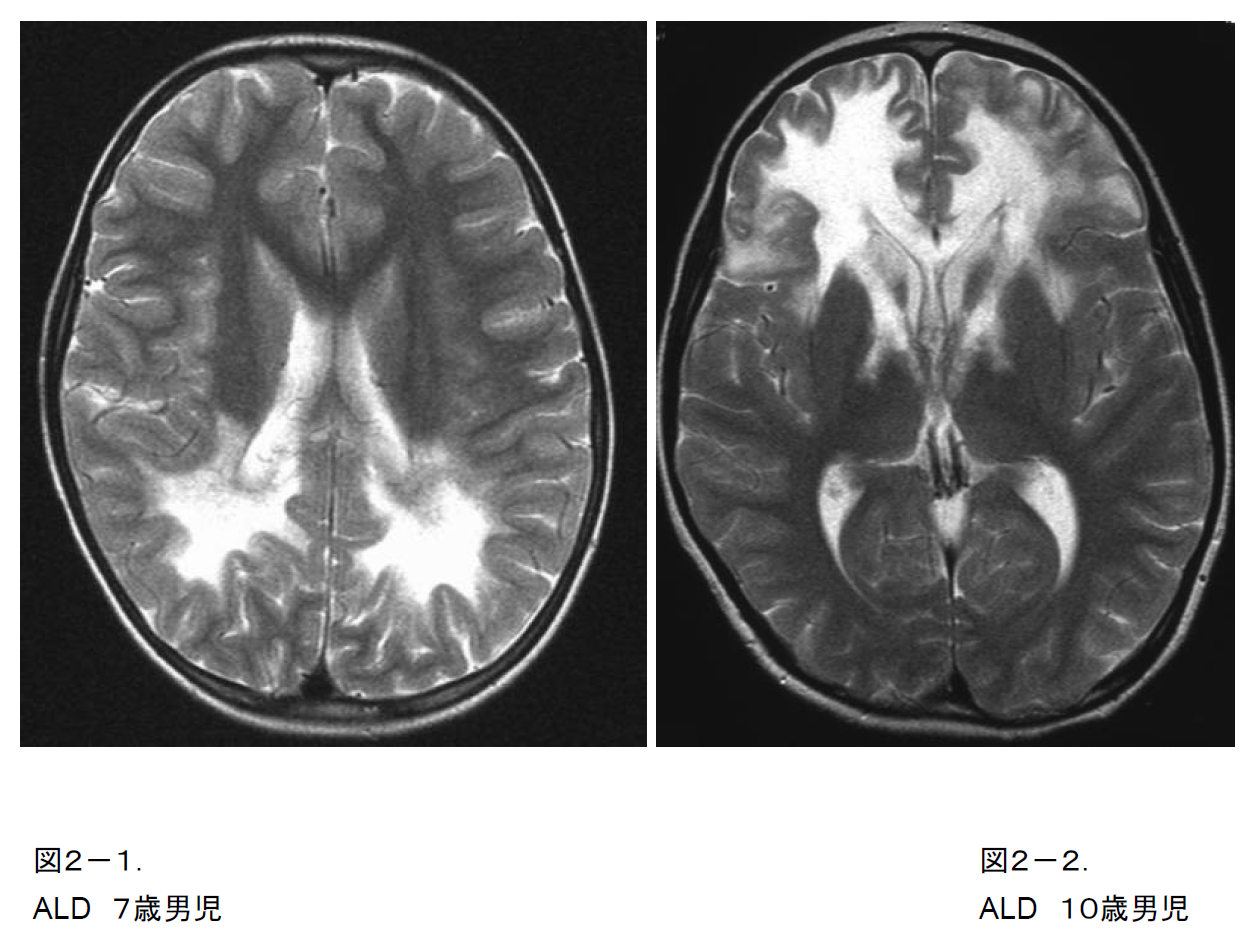
Figure 2-2. T2-weighted image of a 10-year-old boy with ALD. An atypical case starting from the anterior lobe and the anterior limb of the internal capsule.
(c) Krabbe disease
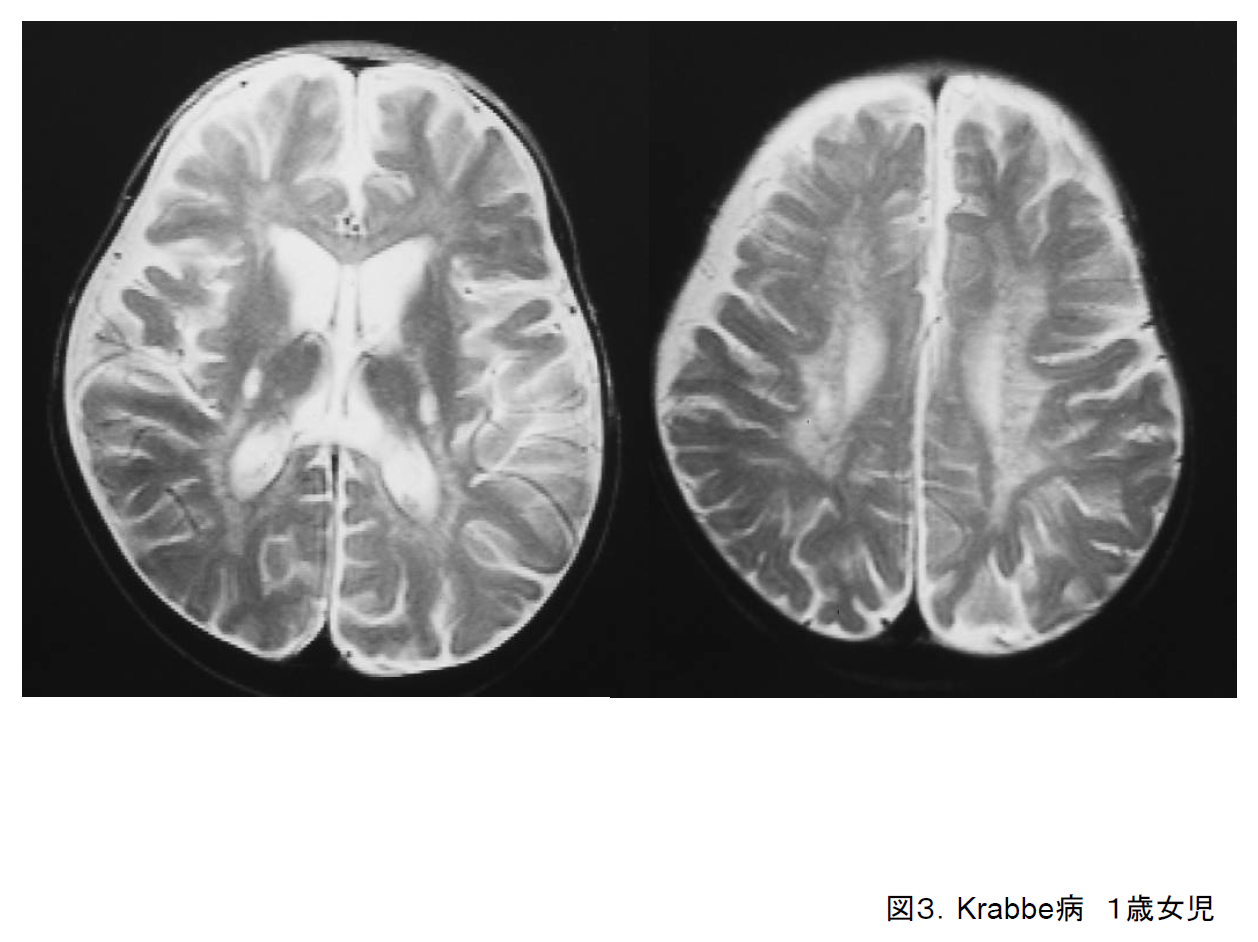
In the infantile form of Krabbe disease (globoid-cell leukodystrophy [GLD]), symptoms such as difficulty in holding up the head, poor feeding, and irritability become noticeable before the age of 6 months. As these symptoms are also seen with peripheral neuropathy, the reduction or loss of deep tendon reflexes is an important characteristic of Krabbe disease. It progresses rapidly until the patient is completely immobile by around 1 year of age. It may also occur later at a wide range of ages, from the late-onset juvenile form to the adult form.
On cranial MRI, demyelination spreads out from the white matter around the posterior horn of the lateral ventricle to the occipital and parietal lobes (Figure 3). Abnormalities of the posterior limb of the internal capsule and the cerebellar medulla are also common.3) Krabbe disease is suspected on the basis of clinical history, imaging, and peripheral nerve conduction tests. The final diagnosis is reached by confirming low galactocerebrosidase activity and a mutation in the GALC gene.
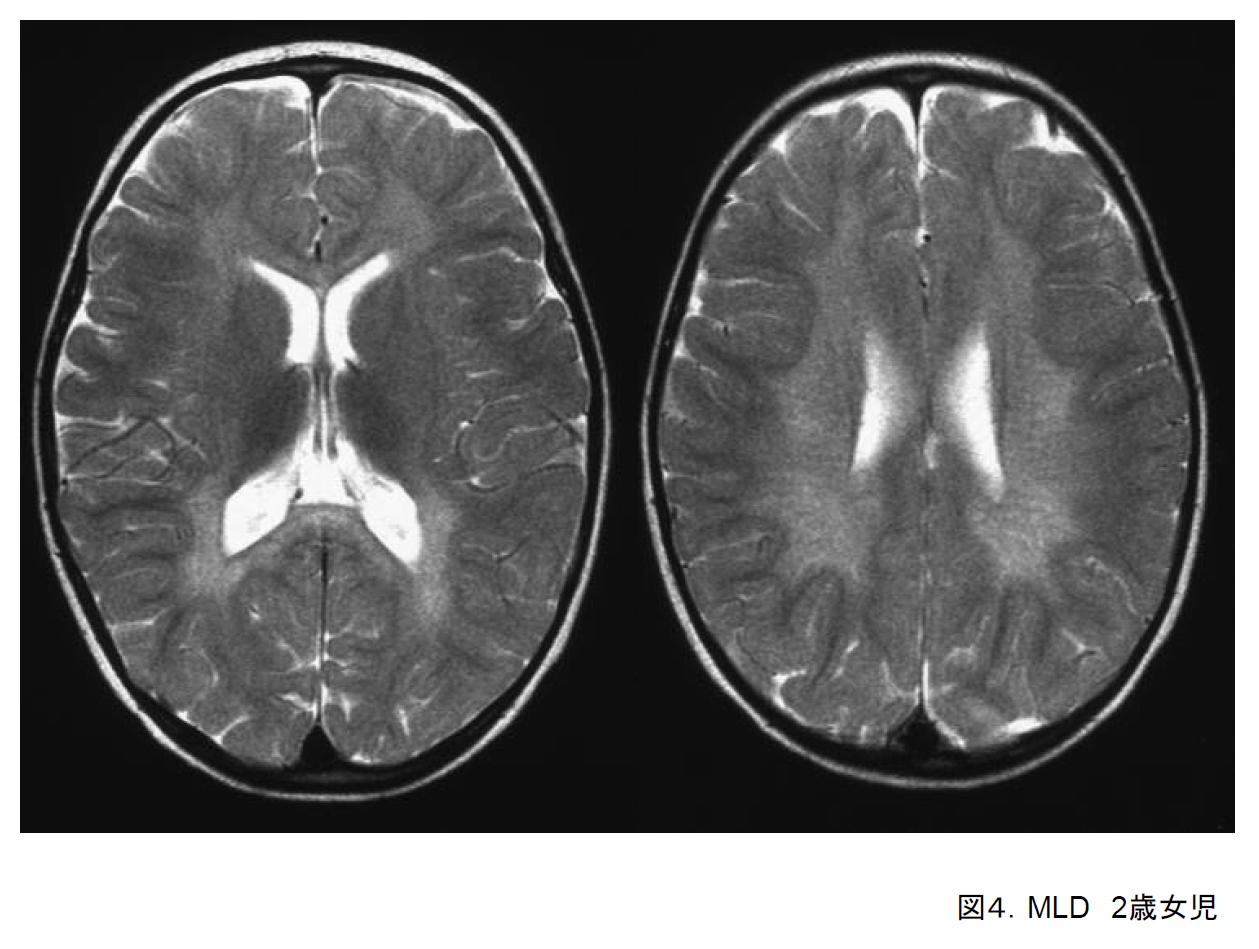
(d) Metachromatic leukodystrophy
In the infantile-onset form of metachromatic leukodystrophy (MLD), symptoms such as becoming prone to falling, loss of muscle strength, and using fewer words become noticeable between the ages of 18 months and 2 years. Spastic palsy, tonic seizures, and other symptoms also appear, and within a few years the patient becomes completely immobile. This is another disorder in which deep tendon reflexes are reduced or lost due to peripheral neuropathy. It is categorized as juvenile-onset or adult-onset according to the age at which it first appears.
On cranial MRI, demyelination is seen to spread out from the deep white matter in the frontal, parietal, and occipital lobes (Figure 4). The diagnosis is suspected on the basis of clinical history, cranial imaging findings, and abnormal peripheral nerve conduction tests, and confirmed by low aryl sulfatase A activity and a mutation in the ARSA gene.
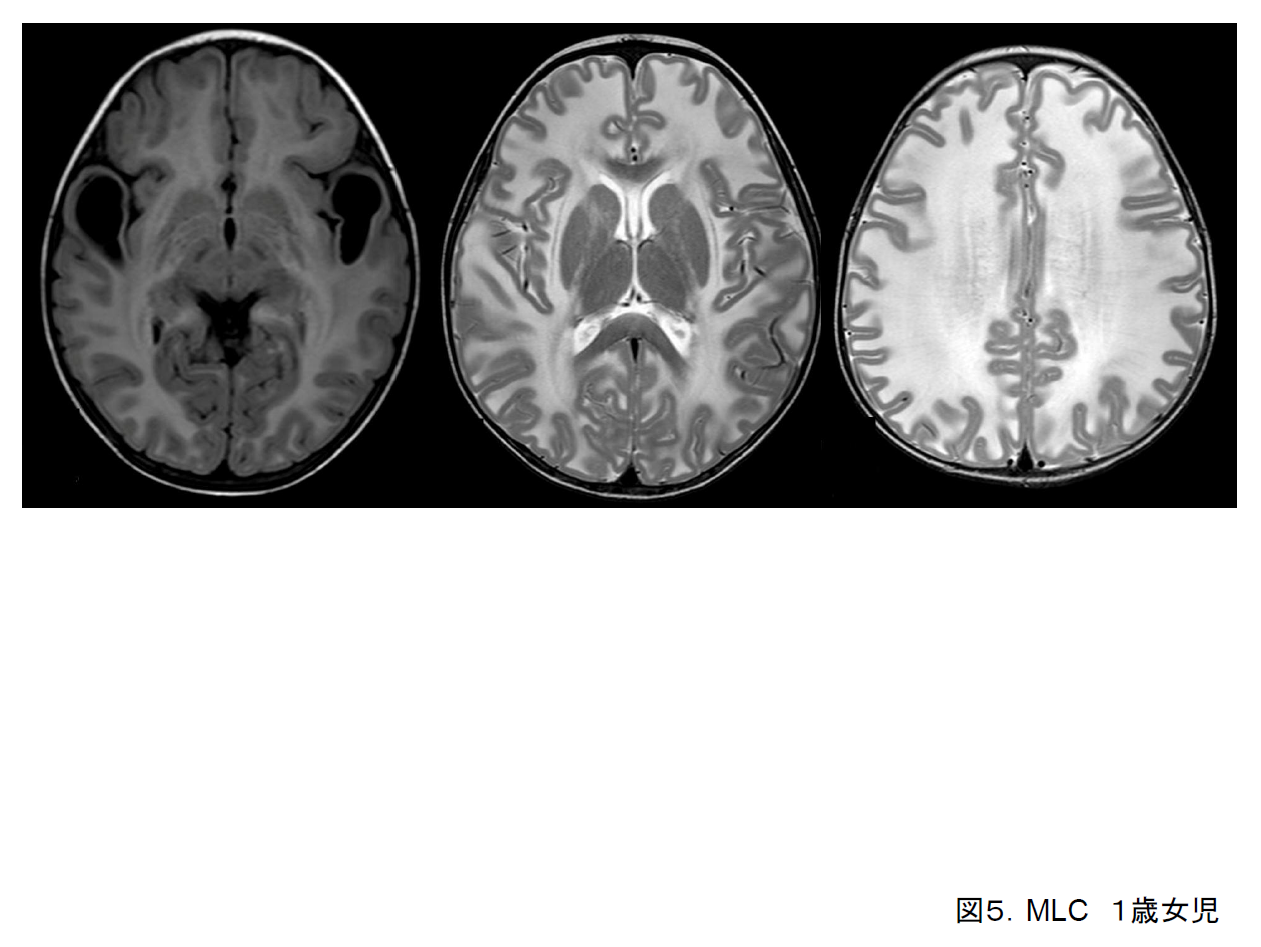
(e) Megalencephalic leukoencephalopathy with subcortical cysts
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) presents early in infancy, and mental and motor development are mildly delayed. Poor muscle tone and unstable gait tend to be present, and seizures may also occur.
Cranial MRI basically reveals an overall increase in the volume of cerebral white matter and hyperintensities on T2-weighted imaging; cysts in the anterior parietal lobe or the subcortical white matter in the frontal and temporal lobes may also be present (Figure 5). This disorder is regarded as a separate disease entity on the basis of cranial imaging findings, and the responsible gene (MLC1) has also been identified.4)
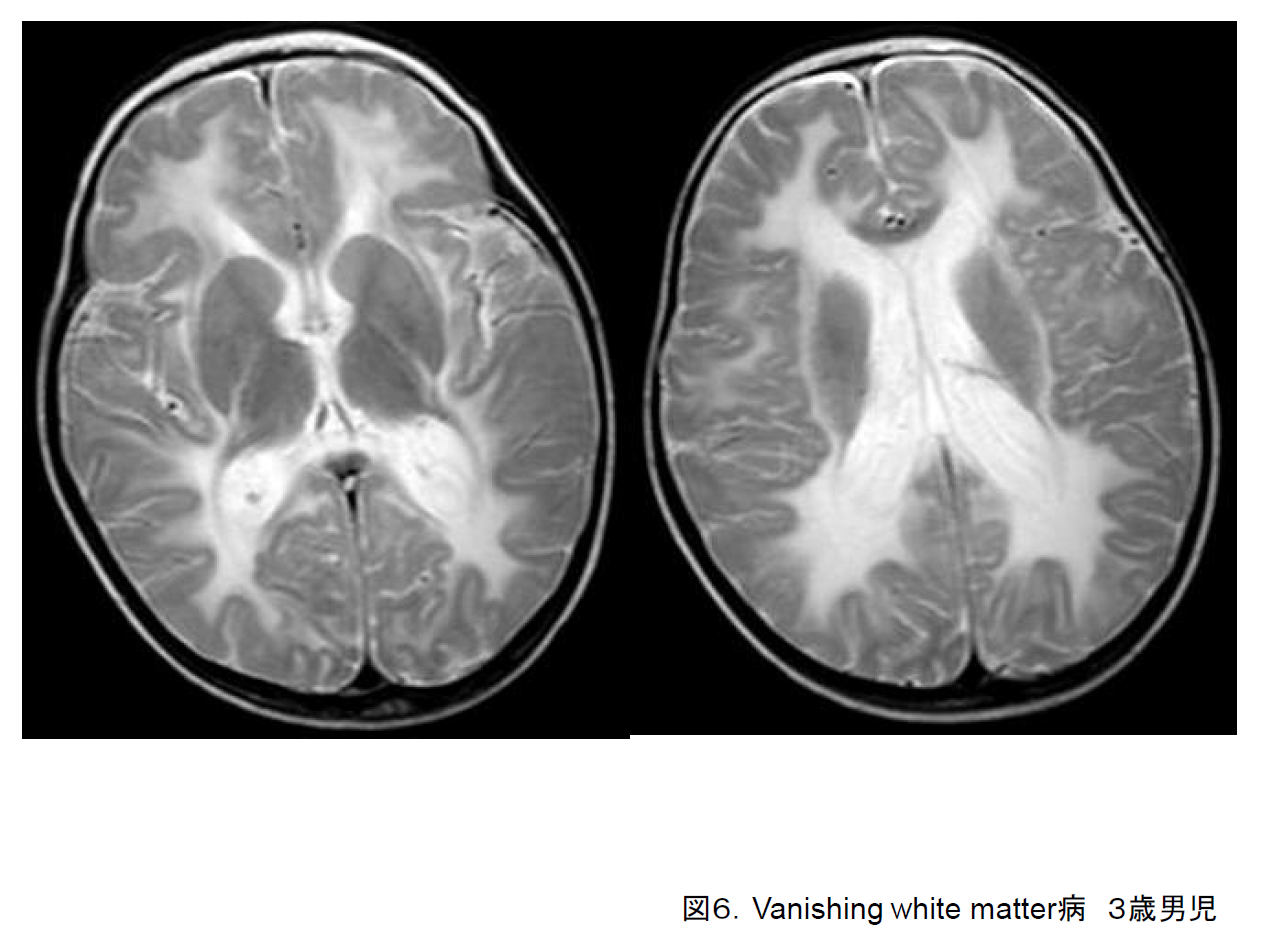
(f) Leukoencephalopathy with vanishing white matter
Also known as vanishing white matter disease (VWM), this disorder may develop at any age from 1 year old to adulthood. In many cases it progresses unevenly, with increased motor or cognitive regression triggered by an infection or minor cranial trauma.5) Cerebral white matter that is hyperintense on T2-weighted imaging (Figure 6) gradually becomes hypointense on FLAIR imaging until it comes to resemble cerebrospinal fluid. Abnormalities of the EIF2B1-5 gene have been reported in cases of VWM.
B.Hypomyelination
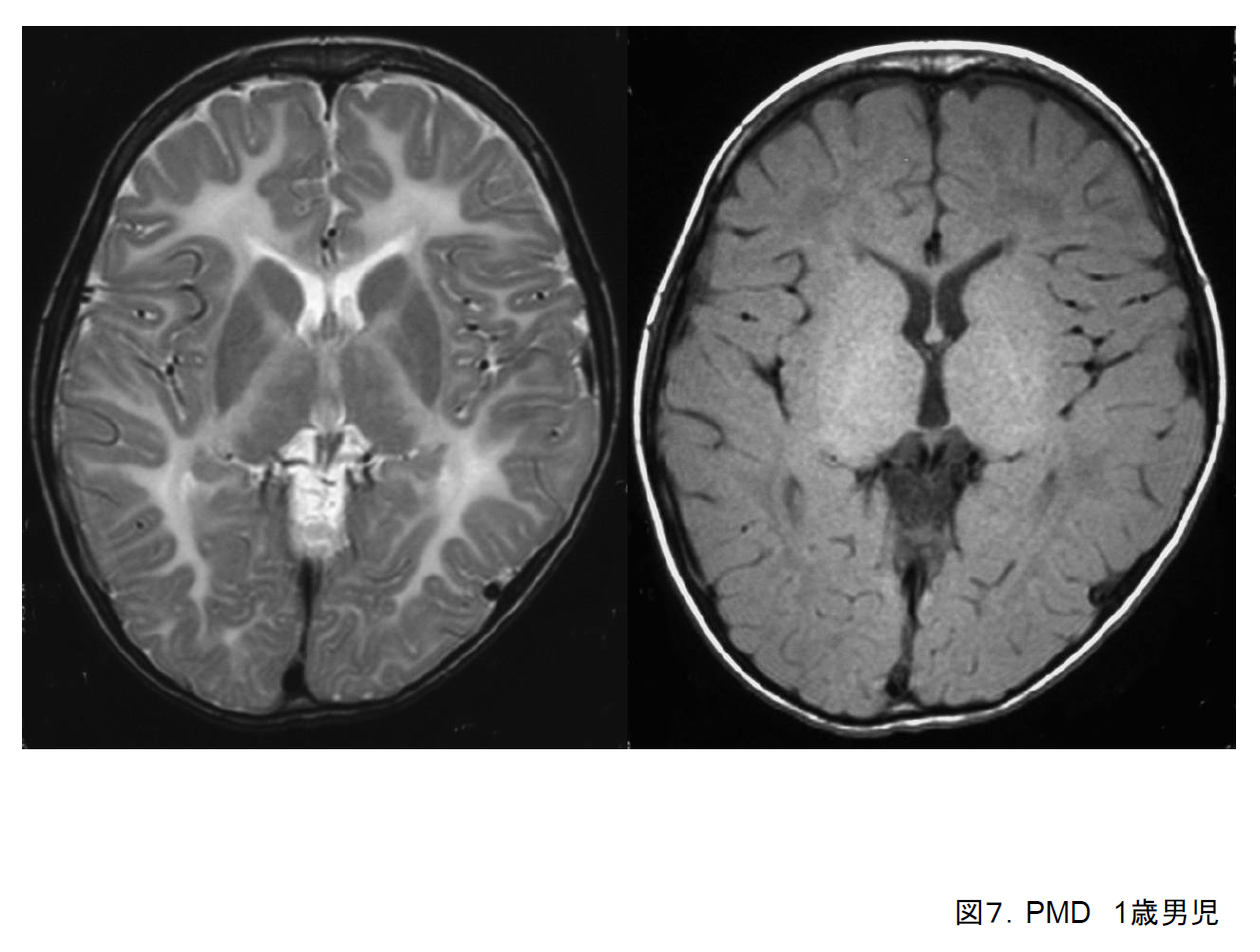
(a) Pelizaeus-Merzbacher disease (PMD)
Symptoms of Pelizaeus-Merzbacher disease (PMD) become noticeable in infancy, including nystagmus (oscillating or rotatory), psychomotor retardation, and poor muscle tone. Cerebellar symptoms (e.g., ataxia, nystagmus), basal ganglia symptoms (e.g., rigidity, dystonia), and tonic seizures may also be present. Some degree of correspondence between genotype and clinical form is known. Babies with the connatal form are unable to hold their heads up and may die during infancy. In the classic form, they are very slow to be able to hold their heads up, but once this is achieved may live to a comparatively advanced age (20–60 years).
On cranial MRI, in the connatal form, the myelin sheaths are almost entirely hyperintense on T2-weighted imaging, indicating that they have hardly formed at all (Figure 7). They are usually isointense on T1-weighted imaging. In some cases, weak hyperintensities suggesting myelin sheath formation may be present.
PMD is caused by duplication or mutation of the PLP1 gene on the X-chromosome and demonstrates sex-linked recessive inheritance. In boys, it should be strongly suspected if nystagmus, hypomyelination on cranial imaging, and waves III and above are lost from the auditory brainstem response (ABR).
(b) Pelizaeus-Merzbacher-like disease
Some patients who exhibit nystagmus and psychomotor retardation from infancy and whose MRI findings also indicate a clinical diagnosis of PMD do not have any abnormality of the PLP1 gene. They include some girls. This PMD-like disorder demonstrates autosomal recessive inheritance, and an abnormality of the GJC2 gene has been identified.
(c) 18q deletion syndrome
Psychomotor retardation, poor muscle tone, short stature, and other symptoms become noticeable in infancy. Facial dysmorphism (including midface hypoplasia, sunken eyeballs, blepharophimosis, and carp-like mouth) may be present, but there are no specific symptoms.
Cranial MRI findings vary widely, from hyperintensity of the cerebral white matter as a whole on T2-weighted imaging to scattered areas of hyperintensity, making diagnosis on the basis of imaging findings difficult.
The diagnosis is made by confirming the deletion of the long arm of chromosome 18 at the G-band. The abnormal myelination is believed to be the result of haploinsufficiency of the myelin base protein (MBP) gene.
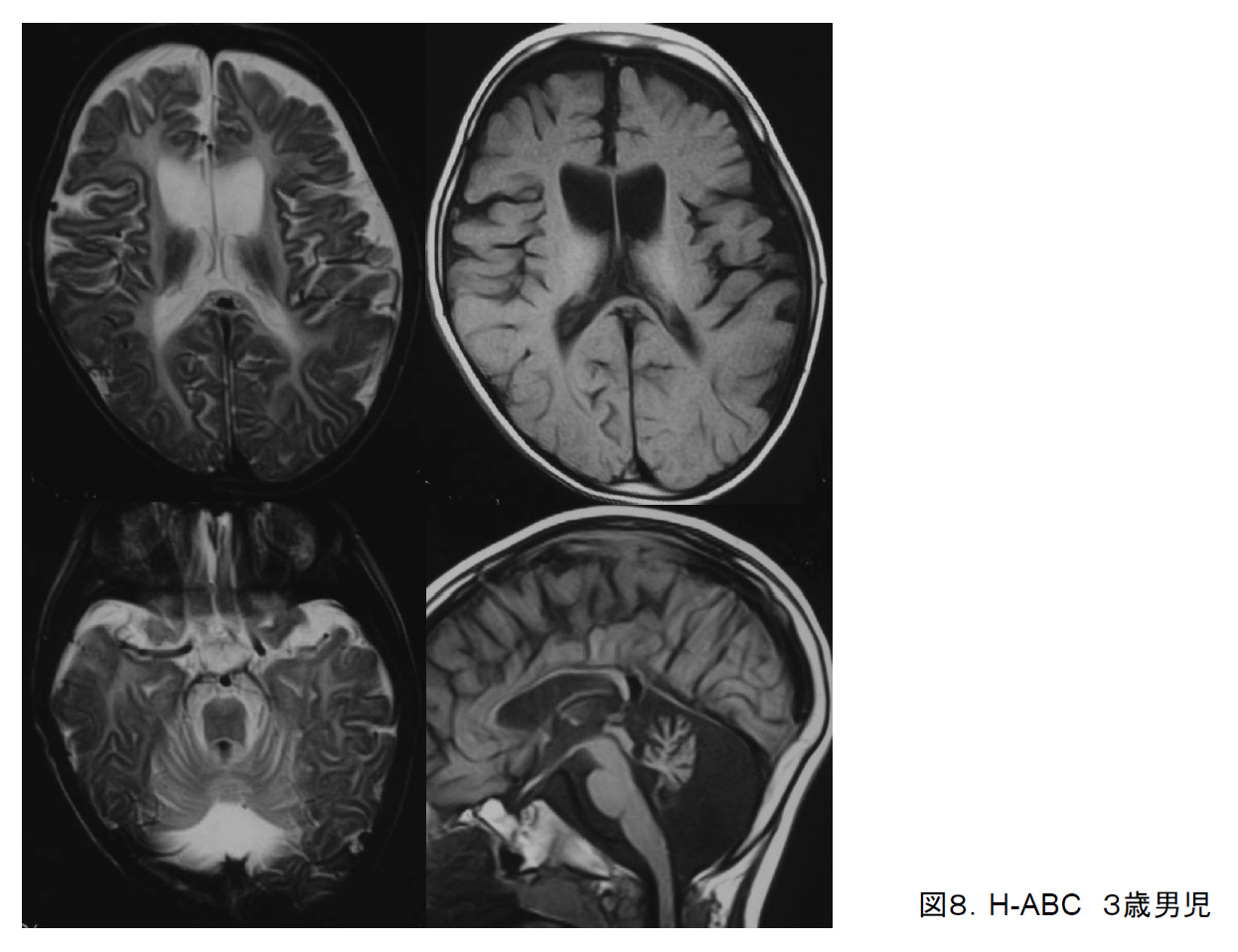
(d) Hypomyelination with atrophy of basal ganglia and cerebellum
Symptoms of hypomyelination with atrophy of basal ganglia and cerebellum (H-ABC) include gait disturbance, dystonia, and cerebral ataxia. They first appear in infancy and gradually progress. The early appearance of dystonia is characteristic. H-ABC has been reported to constitute a separate disease entity on the basis that imaging findings demonstrate atrophy of the bilateral basal ganglia and cerebellum in addition to cerebral white matter hypoplasia on imaging (Figure 8); the responsible gene (TUBB4A) has been identified, and the disease is known to be limited to a specific mutation (D249N).6) It is characterized by prominent atrophy of the basal ganglia, particularly the caudate nucleus and the putamen, visualized as hyperintensities on T2-weighted imaging, and when both this and hypomyelination are present then H-ABC is not difficult to diagnose.
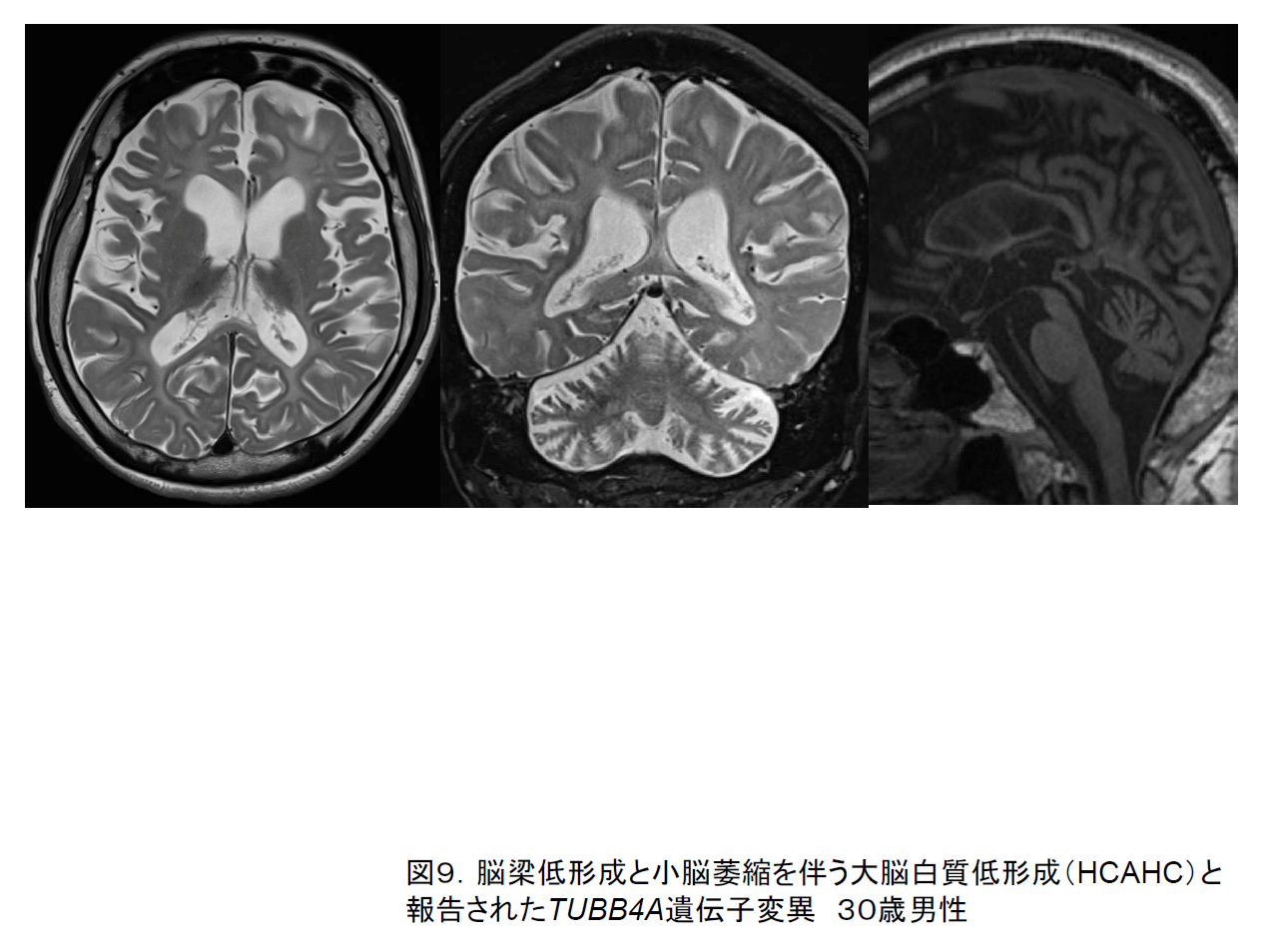
Diseases due to mutations in the TUBB4A gene other than the one responsible for H‑ABC have also been discovered; in one type, although cerebellar atrophy is present, the putamen does not atrophy and its signal intensity is unchanged (Figure 9).7) In another (DYT4) patients only display involuntary movement, with no abnormalities evident on cranial MRI. It has been proposed that these diseases be classed together as TUBB4A-associated disorders.
(e) Pol III-related leukodystrophies
Even when overall cerebral white matter hypoplasia is evident on cranial MRI, it is very often difficult to assign a specific diagnosis. One subset of such disorders comprises those in which a combination of cerebellar atrophy and hypoplasia of the corpus callosum are present. Their basic symptoms comprise progressive gait disturbance due to spasticity and cerebellar ataxia as well as nystagmus, and they have been reported under various names according to their combination with other symptoms, such as abnormal teeth (hypodontia or late eruption) and pituitary hypogonadism (delayed or absent sexual maturation). These include hypomyelination, hypodontia, and hypogonadotropic hypogonadism (4H) syndrome;8) ataxia, delayed dentition, and hypomyelination (ADDH); tremor-ataxia with central hypomyelination (TACH); leukodystrophy with oligodontia (LO); and hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum (HCAHC).9) In all cases, symptoms become evident at age 1–2 years, progression is gradual, and patients become able to walk, albeit unsteadily. The responsible genes (POLR3A/POLR3B) have been identified,10, 11, 12) and as these disorders have been shown to constitute a series of different forms of expression it has been proposed that they be classified as POLIII-related leukodystrophies.13)
On cranial MRI imaging, these disorders all exhibit faint overall hyperintensity of the cerebral white matter on T2-weighted imaging in addition to cerebellar atrophy and thinning of the corpus callosum, with no marked atrophy of the basal ganglia (Figure 10). On T1-weighted imaging the white matter often appears hyperintense, indicating the presence of some degree of myelination. As time passes, there is no increase in myelination on T2-weighted imaging, while myelination is actually lost on T1-weighted imaging.
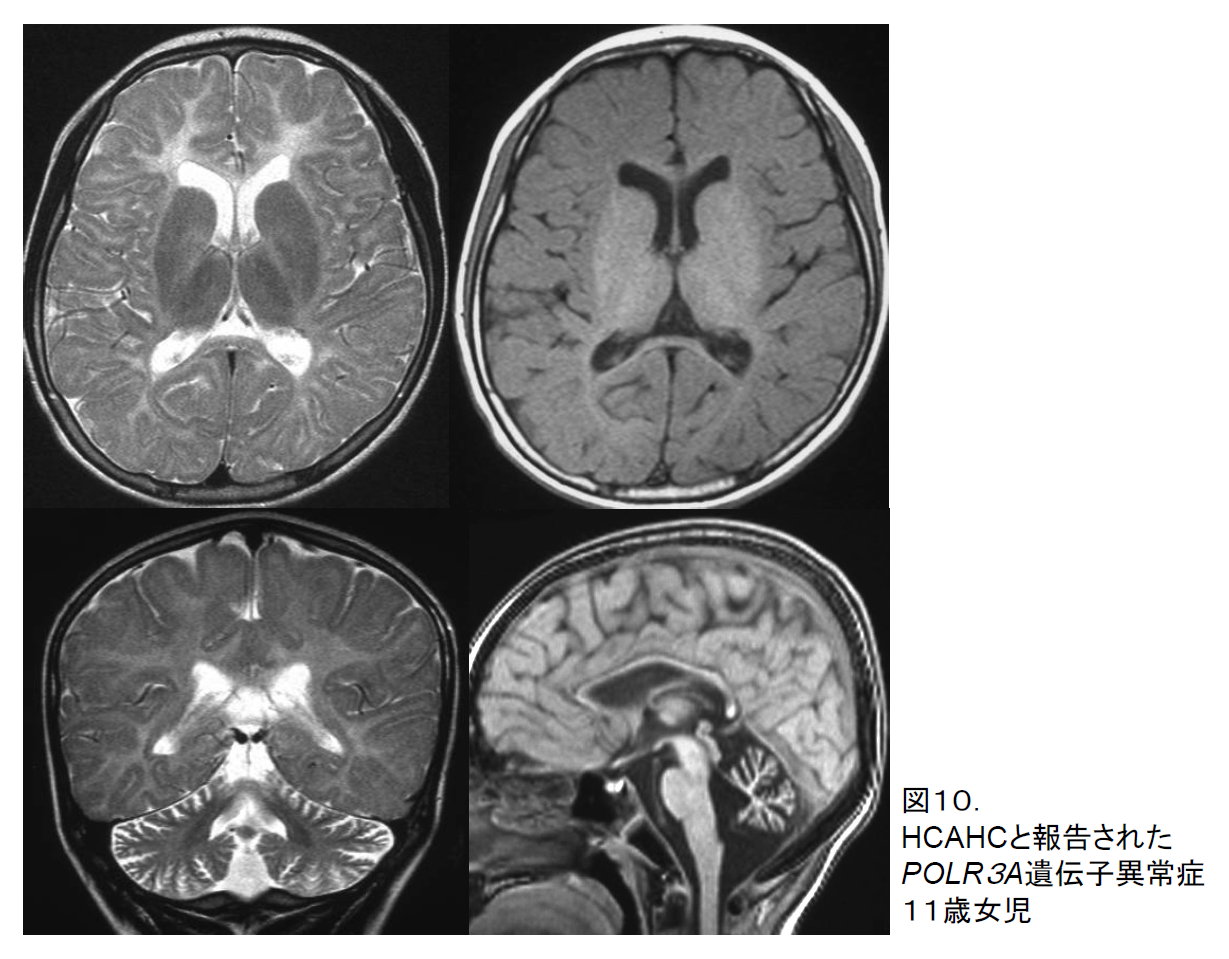
Table 2. Symptoms specific to certain disorders, including hypomyelination
Easily noticeable basic symptoms: poor muscle tone, spastic palsy of the arms and legs, motor retardation, cognitive impairment (ranging from retardation to regression), extrapyramidal symptoms, nystagmus, peripheral neuropathy
| External malformation | 18q deletion syndrome |
| Infantile nystagmus | PMD, PMD-like disease |
| Early dystonia | H-ABC |
| Congenital cataracts | HCC |
| Cerebellar ataxic symptoms | POLIII-related leukodystrophy、H-ABC |
| Endocrine disturbance | POLIII-related leukodystrophy |
| Dental abnormalities | POLIII-related leukodystrophy |
| Myopia | POLIII-related leukodystrophy |
6. References
- Schiffmann R, van der Knaap MS. An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72:750-759.
- Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010;133:2971-2982.
- Sasaki M, Hanaoka S, Takashima S, et al. MRI and CT findings in Krabbe disease. Pediatr Neurol 1991;7:283-288.
- van der Knaap MS, Boor I, Estévez R. Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in bran ion and water homoeostasis. Lancet Neurol 2012;11:973-985.
- van der Knaap MS, Pronk JC, Sheper GC. Vanishing white matter disease. Lancet Neurol 2006;5:413-423.
- Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum.Am J Hum Genet. 2013;92:767-73.
- Miyatake S, Osaka H, Shiina M, et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies.Neurology 2014;82(24):2230-7.
- Timmons M, Tsokos M, Asab MA, et al. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology 2006;67:2066-2069.
- Sasaki M, Takanashi J, Tada H, et al. Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev 2009;31:582-587.
- Saitsu H, Osaka H, Sasaki M, et al. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet 2011;89:644-551.
- Bernard G, Chouery F, Putorti ML, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:415-423.
- Tétreault M, Choquet K, Orcesi S, et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:652-655.
- Bernard G, Vanderver A. Pol III-Related Leukodystrophies. GeneReviews™ [Internet]. In: Pagon RA, et al. ed. Seattle (WA): University of Washington, Seattle; 1993-2013. 2012 Aug 02.