




先天性大脳白質形成不全症の診断基準
(平成23年1月9日)
定義;大脳をはじめとする中枢神経系の白質の髄鞘形成が遺伝的要因により先天的に不完全(低形成)な疾患群と定義される.これは髄鞘の構成成分や髄鞘化に必要な因子などの遺伝的な異常が原因でおこる,中枢神経系の髄鞘化の広範かつ著明な低下あるいは停止を特徴とする疾患群である.現在までにPMDをはじめ,11疾患が知られている(表1).脱髄性疾患および代謝性や全身性障害に伴う二次的な髄鞘化障害による疾患は除外される.
1.共通して認められる臨床症状および検査所見
先天性大脳白質形成不全症に認められる必須所見として,以下の2点がある.
(1)錐体路障害:痙性四肢(下肢)麻痺,(2)MRI画像所見:T2強調画像で,白質にび漫性の高信号領域.脱髄の所見は除外.
また,随伴所見として,以下の6点が挙げられる.
(1)眼振,(2)精神運動発達遅滞,(3)小脳障害:体幹・四肢の失調症状,企図振戦,小児期には測定障害,変換障害,不明瞭言語など,(4)大脳基底核障害:固縮,ジストニア,(5)てんかん,(6)電気生理学的検査所見:誘発電位では中枢伝導障害を示す.
これらの臨床症状の特徴として,錐体路障害は,幼児期には低緊張のことが多いが,上下肢の腱反射が亢進し,Babinski反射等の病的反射が残存し,次第に痙性四肢(下肢)麻痺を呈する.複合型あるいは単純型痙性対麻痺の表現型をもつ軽症例の存在が知られており,下肢痙性が初発症状のことがある.精神運動発達遅滞はほとんどの症例でみられる.一般的に運動障害が,知的障害よりも高度であり,言語理解力が表出言語能力を上回る.小脳障害は遠心路,求心路ともに起こる.体幹・四肢の失調症状,企図振戦,小児期には測定障害,変換障害,不明瞭言語などが認められることがある.固縮やジストニアなどの大脳基底核症状は,経過にともなって出現する場合も多い.白質異常により,皮質症状としてのてんかんを合併することもある.生理学的検査では,聴性脳幹反応や体性感覚誘発電位,視覚誘発電位では中枢性伝導障害を示す.また,末梢神経伝導速度あるいは筋電図所見により末梢神経障害がある場合はI群,ない場合はII群に分類する.画像検査では,MRI撮影が診断的に有用かつ必須である.T2強調画像で,白質にびまん性の高信号領域を認める.CT像では,白質の低吸収域を呈することがあるが,診断の有用性は低い.血清・生化学的所見では,Allan-Herndon-Dudley症候群(AHDS)を除き,特異的な所見はない.
2.鑑別診断のためのフローチャート(図1)
まずMRI所見より,T2強調画像での高信号が正常の髄鞘化の遅延・停止なのか脱髄なのかを明らかにする.軽度の髄鞘化遅延は,半年以上の経過をおいて再検査し,髄鞘化遅延の有無を確定する.脱髄性疾患では,T2強調画像で著しい高信号を呈する部位を認めることが多く,同部位はT1強調画像では低信号を呈することがある.髄鞘化遅延の判断には,正常小児の髄鞘化パターンを知ることが必要である.ついで末梢神経伝導速度測定などにより末梢神経障害のない群(1群)と末梢神経障害を合併する群(2群)に大別する.各群に含まれる疾患は表1に示す.
次に遺伝学的な検査を念頭に鑑別診断を進める(図2).家族歴の有無,発端者の性別,発症年齢,眼振の有無などの臨床情報が重要である.第1群については,まず,多発奇形を伴う場合には,18q欠失症候群を疑い,染色体検査あるいは18qサブテロメアFISHを行う.男児で甲状腺機能異常を認める場合はAHDSを疑う.これらの異常がない先天性大脳白質形成不全症の男児の場合は,最も頻度の高いPMDを疑い,PLP1遺伝子異常の検索を行う.PLP1の異常が認められない男児,あるいは女児の症例では,PMLD1を疑いGJC2遺伝子の異常を検索する.これらで異常が認められない場合は,Salla病(SD), Hsp60 chaperon病(MitCHAP60),基底核および小脳萎縮を伴う髄鞘形成不全症(HABC),小脳萎縮と脳梁低形成を伴うび漫性大脳白質形成不全症(HCAHC)の可能性を疑う.SDでは尿中あるいは髄液中の遊離シアル酸の高値,HABCでは大脳基底核や小脳の萎縮,HCAHCでは大脳基底核萎縮を伴わない小脳萎縮と脳梁低形成が特徴である.一方で,PLP1遺伝子の部分重複やPLP1やGJC2の翻訳領域外の変異など,一般的な遺伝子解析方法では検出が困難な稀な変異によるPMDあるいはPMLD1の可能性も考慮する.
第2群については,随伴する症状の特徴から,比較的容易に鑑別が可能である.白内障を伴う場合には先天性白内障を伴う髄鞘形成不全症(HCC),歯牙低形成や成長障害が認められれば失調,歯牙低形成を伴う髄鞘形成不全症(ADDH),Waadenburg症候群やHirschsprung病を認める場合には脱髄型末梢神経炎,中枢性髄鞘形成不全症,Waadenburg症候群,Hirschsprung病(PCWH)と考えられる.なお,PLP1遺伝子機能喪失変異によるPMD患者は,末梢神経障害を呈する.
本分類基準のいずれの疾患にも該当しない症例が少なからず存在することから,まだ疾患概念が確立されていない先天性大脳白質形成不全症があると考えられる.
3.各疾患の概要
第1群;主として中枢神経の先天性髄鞘形成不全症
(1)Pelizaeus-Merzbacher病
(Pelizaeus-Merzbacher disease;PMD,HLD1,OMIM#312080)
疾患の概念
X連鎖性劣性遺伝形式をとる中枢神経系の髄鞘形成不全症であるため,患者は例外を除き男児のみである.神経病理学的には,中枢神経系白質の髄鞘が消失・あるいは極度に低下するが,神経細胞および軸索は保持され,明らかな脱髄性変化を伴わず,残存した髄鞘が斑状のtigroidの像を呈することを特徴とする.遺伝学的には,プロテオリピドプロテイン1(proteolipid protein 1; PLP1)遺伝子の異常がこの疾患の主たる原因である1).
臨床症状
生直後から遅くも1カ月程度までに眼振で気づかれることが多い.水平性眼振であることが多く,代償性の頭部振戦を認める場合がある.生後から半年程度までは筋緊張低下の症状を呈するが,原始反射の消失が遅れ,Babinski反射は半年を超えても陽性であり,やがて腱反射の亢進も明らかになり一次ニューロンの問題を示す.小脳症状としての企図振戦は1歳過ぎには,注意深く観察すると明らかであることが多い.また,2歳頃にはアテトーゼ様の異常肢位が発現してくる.このように中枢神経系の,運動,運動制御系,大脳基底核のすべての症状が相次いで出現するのがこの疾患の特徴であるが,後年眼振は目立たなくなり,関節拘縮が進むと小脳症状も気づかれず,年長児で痙性と固縮をもつ脳性麻痺として診断されている例も多い.また,乳幼児期の運動知的発達が正常で,学童期以降にゆっくりと退行する,X連鎖性痙性対麻痺の表現型をとることもある.通常10〜20歳代を過ぎると症状の退行が始まり,平均寿命は30歳前後である.症状の退行と平行して,画像上の脳萎縮を認める.PLP1機能喪失変異の症例では,末梢神経障害を呈する.
検査・画像所見
検査所見では,聴性脳幹反応では,II波以降がまだらな中枢伝導時間を反映して消失する.末梢神経伝導速度は正常であるが,PLP1欠失など機能喪失型の変異による症例では,軽度低下する.血清生化学・尿所見に異常は認めない.髄液所見では,蛋白上昇を含め明らかな異常は認めない.脱髄疾患とは異なりmyelin basic proteinの値も正常範囲から正常上限であることが多い.MRI画像は極めて特徴的であり,T2強調画像ではび漫性の白質の高信号およびT1強調画像で,皮質・白質のコントラストが消失(あるいは低下)していることが特徴的である.一般的に,T2強調像の信号では正常の新生児期よりも髄鞘化に乏しく年齢が長じても変化が乏しい.T1強調画像上は,ゆっくりとした髄鞘化を認めるが,皮質・白質のコントラストが悪い.CT検査では,非特異的な大脳萎縮を認めるのみで診断的な情報は少ない.
遺伝子診断
PLP1遺伝子重複,点変異,遺伝子欠失など様々な変異が存在する.1)PLP1遺伝子重複はほぼ半数(50-75%)の症例で認める.これは,定量的PCR法や間期核FISH法などにより正常の2倍量のPLP1の存在により確認できる.また,新たな技術としてMLPA(multiplex ligation-dependent probe amplification)やアレイCGH(microarray-based comparative genomic hybridization)などでも診断可能である.2割前後(15-25%)の患者では,PLP1タンパクコード領域やスプライス部位の点変異による.アミノ酸置換型変異が最も頻度が高いが,他にスプライシング異常やナンセンス変異,挿入あるいは欠失なども認める.変異の検出には,直接塩基配列決定法などを用いる.PLP1遺伝子全長の欠失は稀である(2%以下).欠失は上記のいずれの方法でも検出される.PMD遺伝子診断は,変異の多様性を念頭に置き,異なる検査方法を組み合わせる必要がある.
(2)Pelizaeus-Merzbacher様病1
(Pelizaeus-Merzbacher like disease 1;PMLD1,HLD2,OMIM#311601)
疾患概念
臨床的にはPMDと区別がつかない症状を呈するにもかかわらず,PLP1遺伝子の異常を認めない症例をPMLDと呼び,PMDと区別する.従来,PMD女児例として報告されたいたものを含む.稀な疾患である.常染色体劣性遺伝形式をとる一部の症例では,gap junction protein C2(GJC2)遺伝子の変異が原因であり4),これをPMLD1と呼ぶ.
臨床症状と検査所見
生後早期より眼振を認め,1歳前に運動発達遅滞に気づかれ,その後PMDと同様に錐体路症状,小脳症状,大脳基底核症状が明らかになる.また,聴性脳幹反応では中枢伝導時間の延長を示し,頭部MRIのT2強調画像では白質のび漫性高信号などの所見を呈する.これらの臨床症状や検査所見はPMDに酷似する.一方で,33例の報告例の内12例で自立歩行が可能であると報告されておりPMDより最大運動発達は良好である.しかしながら全例で10歳前に退行が始まり,10歳までには支持歩行を失う.痙性下肢麻痺の経過をとる軽症例の報告もある.
原因遺伝子
GJC2遺伝子(別名Cx47あるいはGJA12)の異常が2004年に報告され,遺伝学的にもPMDとは異質であることが明らかとなった4).その一方で,GJC2変異のないPMLD患者も存在することから,これ以外の遺伝子の関与も示唆されている.既知の変異は,すべてコード領域内の変異であるので,遺伝子診断では,塩基配列決定法などを用いて変異を検出する.これらの変異は,オリゴデンドロサイトの細胞膜上のgap junctionの機能を喪失させると考えられている5).
(3)基底核および小脳萎縮を伴う髄鞘形成不全症
(Hypomyelination with atrophy of the basal ganglia and cerebellum;HABC,HLD6,OMIM%612438)
疾患概念
先天性の白質形成不全に加えて,大脳基底核(尾状核頭,および被殻に顕著)の進行性萎縮を認める疾患.HABCは,2002年に初めて症例報告され,2007年に疾患概念として確立された新しい疾患である6, 7).血族婚例があることより,常染色体劣性が疑われるが,原因遺伝子は未同定である
臨床症状
生下時には異常なく,眼振も認めない症例が多い.最大運動機能としては,支持歩行を3歳頃までに獲得する.10歳前に発達退行を示し数年の経過で支持歩行を消失する.生下時にははっきりしないが,徐々に痙性,小脳失調を示し,錐体外路症状(アテトーゼ,ジストニア,固縮)が出現するのが特徴である.精神遅滞は,中等度から重度だが,発語がなくとも笑顔など非言語性のコミュニケーションが可能な例がある.半数近くの症例で感音性難聴や小頭症,低身長をみる.
検査所見
頭部MRIではびまん性のT2強調高信号を呈する.脳幹は低信号だが錐体路が高信号として描出されるのが特徴である.白質容量は低下して脳室拡大を認める.大脳および小脳白質に認めるT2の高信号は徐々に低信号化し,ミエリンの消失を反映する.小脳,特に虫部の萎縮性変化が著しい.視床・淡蒼球は保たれるが被殻は小さく,やがて消失する.尾状核も徐々に萎縮する.
原因遺伝子
原因遺伝子は未同定である.
(4)18q欠失症候群
(Chromosome 18q deletion syndrome;18qdel,OMIM#601808)
疾患概念
18q欠失症候群は,18番染色体長腕端部欠失による染色体異常症候群で,欠失範囲に多数の遺伝子が含まれるため,多様な症状をきたす.大脳白質形成不全症はmyelin basic protein (MBP)遺伝子の欠失が原因である8).
臨床症状
成長障害(特に低身長),発達遅滞,筋緊張低下,協調運動障害,眼振,伝音性難聴,けいれん,小頭症,顔面正中部低形成,くぼんだ眼球,眼裂狭小,鯉様の口などの多様な臨床症状を呈する.生命予後は良好である.18q欠失症候群の重要な合併症として,膀胱尿管逆流,尿路感染症があげられる.
検査所見
頭部MRIではT2強調画像にて大脳白質の高信号を認める.大脳白質全体の高信号から散在性の高信号までその程度は様々である.脳梁の低信号は保持されやすい.血液検査において IgA 欠損や成長ホルモン分泌低下がしばしば認められる.末梢神経伝導速度,視覚誘発電位は正常との報告がある.聴性脳幹反応に関しては報告が見あたらない.
遺伝子診断
18q21→qterの長腕端部欠失の報告が多く,症例の3/4はde novo変異(発端者突然変異)である.他に家族性の相互均衡転座,de novo転座,家族性の逆位などの報告がある.診断はG分染法染色体検査およびFISH法を用いる.症状に関与する染色体責任部位は18q22→q23とされる.大脳白質病変を呈する症例では,FISH解析で18q23に位置するMBP遺伝子の1コピー欠失を同定する.髄鞘化遅延はMBP遺伝子のハプロ不全によりおこると推測される.
(5)Allan-Herndon-Dudley症候群
(Allan-Herndon-Dudley syndrome;AHDS,OMIM#300523)
疾患概念
X連鎖精神遅滞症候群の1つである.重度の精神発達遅滞,構音障害,アテトーゼ運動,筋低形成,痙性対麻痺を主症状とし,乳幼児期からMRI検査により髄鞘化の遅れが認められる.Xq13.2に存在するsolute carrier family 16, member 2 (SLC16A2; monocarboxylic acid transporter 8 (MCT8))遺伝子の異常による甲状腺ホルモンの膜輸送の障害が原因と考えられている9).近年,PLP1などの既知遺伝子異常を認めないPelizaeus-Merzbacher病患者の約1割がSLC16A2 (MCT8)の変異を有することが報告された10).
臨床症状
いわゆる症候性X連鎖精神発達遅滞を示すが,幅広い症状のスペクトラムを示す.最重症例においては,著しい低緊張,眼振,ジストニア,強剛痙性対麻痺を示し,運動や言語の発達遅滞を示す.軽症例は男性にも女性にも認められ,筋緊張低下から次第に痙性対麻痺や構音障害,失調歩行,舞踏病運動,顔面筋罹患を来す.甲状腺ホルモン以外の内分泌機能に異常はなく,内臓奇形などは合併しない.難聴も認められない.
検査・画像所見
甲状腺ホルモンの異常と特徴的な画像所見を示す.甲状腺ホルモン検査ではT4低値,T3高値を示し,TSHは正常値の上限を示すことが多い.頭部MRI検査では初期にはほとんど異常が見られないこともあるが,次第に皮質萎縮,あるいはT2強調画像でのび漫性高信号などの髄鞘低形成の所見を示す.文献的に聴性脳幹反応の報告はないが,中枢神経における伝達速度が低下するとされている.
遺伝子診断
SLC16A2 (MCT8)の変異を塩基配列決定などで同定する.SLC16A2 はX染色体q13.2に位置し,12膜貫通ドメインを持つタンパクをコードしている.SLC16A2 (MCT8)は甲状腺ホルモンの膜能動輸送に関わっていると考えられている.ナンセンス変異やミスセンス変異が報告されており,変異により重症度が異なる.SLC16A2 (MCT8)領域の転座による女児例の報告もある.以下のような症状を示す場合,SLC16A2 (MCT8)変異を疑う.①重度精神運動発達遅滞を伴うX連鎖痙性対麻痺,②頭部MRI画像で髄鞘形成障害を認める,③甲状腺ホルモン検査でT4低値,T3高値,TSHは正常値の上限を示す.
(6) HSP60 chaperon病
(Mitochondrial Hsp60 chaperonopathy;MitCHAP60,HLD4,OMIM #612233)
疾患概念
ミトコンドリアシャペロニンHsp60蛋白質の異常による先天性大脳白質形成不全症である.原因遺伝子は2q33.1に位置するHsp60遺伝子(HSPD1)で,常染色体劣性遺伝形式をとる.これまでに報告されたのは,血族結婚を繰り返したイスラエルの大家系のみである11).
臨床症状
上記家系の10名の症例報告によると,生直後〜3カ月より筋緊張低下,眼振,精神運動発達遅滞に気付かれる.顕著な痙性,発達遅滞,退行をみる.てんかんの合併は約半数に見られる.摂食不良による栄養不良ならびに成長障害をみる.20歳までに嚥下性肺炎や原因不明の突然死により死亡する症例が多い.出生後1−2年で死亡する重症例もある.2年以上生存した患者は,進行性の四肢の痙性麻痺と拘縮を認める.家族内・家族間での表現型の差異を認める.
検査所見
MRIで大脳および小脳白質にT2強調画像でび漫性に高信号を呈し,髄鞘化を全く認めない.脳梁の菲薄化と脳室拡大,脳幹と小脳の萎縮も認める.聴性脳幹反応ではI波の遅延とII波以降の消失をみる.
遺伝子診断
HSPD1のDNA塩基配列決定法による.上記の家系ではD29G変異のホモ接合体で発症し,へテロ接合体の保因者は無症状である.HSPD1の他の変異は,常染色体優性遺伝形式をとる痙性対麻痺13型(SPG13;OMIM#605280)の原因となる.
(7)Salla病
(Salla disease;SD,OMIM#604369)
疾患概念
SDは,常染色体劣性遺伝形式をとり,リソゾームへのシアル酸蓄積を特徴とする.MRI所見上,先天性大脳白質形成不全症に共通するび漫性白質病変を呈する.原因遺伝子は,SLC17A5遺伝子 (6q14-q15) である.同じ遺伝子変異による infantile sialic acid storage disorder (ISSD, OMIM#269920) は乳児期に致命的であるが,SDは乳児期に発症するが進行は緩徐であり,軽症型ないし成人型とされる.
臨床症状
多くの症例はフィンランド(Salla地方)に集積している.発達遅滞,発達退行,身体発育不全,運動失調,眼振,筋緊張低下,痙性,てんかんなどを呈する.
検査所見
尿中遊離シアル酸(N-acethylneuraminic acid;NANA)の増加が診断に有用である.まれに尿中NANAの増加を見ない症例も存在するが,髄液中のNANAは全ての症例で増加する.MRIではT2強調画像で白質の広範な高信号 (hypomyelinating pattern)を認め,PMDと近似する.MRS検査ではN-acetylaspartate(NAA)の高値を呈するが,これはNANAとNAAは1.5テスラMR装置では区別できないためであり,実際にはNANAの増加を反映しており,診断的に有用である.末梢神経伝導速度は約半数で低下するが,視覚誘発電位,聴性脳幹反応は通常正常である.
遺伝子診断
SDは,SLC17A5遺伝子のR39Cの創始者異常による症例がほとんどであり,本邦での症例は報告されていない.近年,フィンランド人で見いだされているR39Cの変異以外にも少数のSDの原因遺伝子変異が明らかになっている.変異SLC17A5のグルタミン酸/アスパラギン酸トランポーター機能異常による神経伝達障害が大脳病変の分子病態と推測される.
(8)小脳萎縮と脳梁低形成を伴うび漫性大脳白質形成不全症
(Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum;HCAHC)
疾患概念
2009年に本邦から提唱された新しい疾患概念12).中枢神経系の髄鞘形成不全に加え,小脳萎縮と脳梁低形成を伴う.大脳基底核の萎縮は認めない.
臨床症状
これまで報告された3例はいずれも1〜3歳代に自立歩行を獲得しているが,10代に入り徐々に進行する歩行失調,振戦,緩徐言語,錐体路症状を認め,軽度から中等度の精神運動発達遅滞を示している.アテトーゼを認める例はないが,2/3例でジストニアを呈する.
検査所見
末梢神経伝導速度,視覚誘発電位,聴性脳幹反応は正常である.MRI所見は,び漫性T2高信号とT1での同等あるいは軽度高信号を呈す.脳梁低形成と小脳(ことに皮質)萎縮を示す.当初認めない大脳萎縮も,臨床的な退行が認められる時期には観察される.
遺伝子診断
原因遺伝子は不明であり,家族例の報告もなく遺伝形式も不明である.
(9)その他,分類不能な疾患
第2群;中枢神経および末梢神経の先天性髄鞘形成不全症
(1)先天性白内障を伴う髄鞘形成不全症
(Hypomyelination and congenital cataract;HCC,HLD5,OMIM#610532)
疾患概要
大脳白質形成不全と白内障を合併する1疾患単位として,2006年に初めて報告された常染色体劣性遺伝形式をとる新しい先天性大脳白質形成不全症である13).稀少で,これまで10例の報告があるのみである.
臨床症状
生後1カ月未満に白内障に気づかれ,定頸や座位獲得の異常はないが,1歳過ぎに歩行が未獲得であることより発達遅滞に気づかれる.2歳頃までに支持歩行を獲得するが,独立歩行には至らない.緩弱に退行し,9/10例で成人までに歩行を消失する.全例で錐体路徴候があり,病的反射は陽性であるが腱反射は低下していることが特徴である.9/10例で小脳症状を認めており,また,末梢神経症状として筋力低下と下肢遠位筋の萎縮を認めている.大脳基底核症状(ジストニア,アテトーゼなど)については記載がない.精神発達遅滞は軽度から中等度で,熱性けいれん以外のけいれんは2例で認めている.
検査所見
9/10例で末梢神経伝導速度の低下を認め,末梢神経生検では有髄線維の減少と,電顕上,髄鞘形成不全像が認められている.また,MRI所見は,び漫性T2高信号とT1での同等あるいは軽度高信号を呈すが,一部脳室周囲にT2の強い高信号,T1の低信号領域を認め,このような部位では白質の水分含量が増大している可能性が示されている.
遺伝子診断
疾患原因遺伝子FAM126A(DRCTNNB1A)(7p15.3)のDNA塩基配列決定法により変異を同定する.FAM126Aはβカテニン/Wntシグナルに関連し,オリゴデンドロサイトの分化への関与が示唆されている.
(2)失調,歯牙低形成を伴う髄鞘形成不全症
(Ataxia, delayed dentition, and hypomyelination;ADDH,HLD7,OMIM 612440)
疾患概要
進行性の失調,乏歯・欠歯などの歯牙低形成を特徴とする大脳白質形成不全症である14).発症は,幼児期から小児期早期で症状は全体に進行性である.現在まで報告症例数が20家系足らずの稀な疾患である.原因遺伝子は未同定である.ほとんどが孤発例であるが,血族婚家系での同胞発生もあり,常染色体劣性および常染色体優性遺伝形式いずれの可能性もあり,遺伝的異質性が考えられる.ほかに,精神遅滞,下垂体低ゴナドトロピン性性腺機能低下なども合併することもある.
臨床症状
乳幼児期の発達は正常なことが多い.しかし,独歩達成時期から運動発達の遅れが目立つようになる.幼児期正常発達例でも10代前半までには運動失調が目立つようになり,症状が目立つ年齢にはやや家系ごとに幅がある.錐体路症状の有無は症例ごとに異なる.乏歯・欠歯などの歯牙低形成(主に切歯が欠損)は幼児期から気づかれる.ほかに,筋緊張低下,成長障害なども目立つことがある.下垂体性低ゴナドトロピン性性腺機能不全による二次性徴を認めない例もあるが,この内分泌学的特徴が本症に共通する特徴であるかは,報告症例の年齢の違いが大きいためにはっきりしない.精神発達は,正常から中等度精神遅滞まで幅がある.予後に関しては症例数が少ないため不明である.
検査所見
頭部MRIにて,T2強調画像での大脳白質のびまん性高信号による髄鞘低形成と小脳の萎縮を認める.脳梁は菲薄化する.MRSでの診断も有用で,大脳白質におけるmyo-inositolの上昇と,NAAおよびcholineの低下を特徴とする.歯牙低形成は,パノラマ撮影で切歯の欠損を確認する.
遺伝子診断
原因遺伝子および遺伝子座は未同定である.
(3)脱髄型末梢神経炎,中枢性髄鞘形成不全症,Waardenburg症候群,Hirschsprung病
(Peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease;PCWH,OMIM#609136)
疾患概要
オリゴデンドロサイトの発生異常に加え,Schwann細胞,メラノサイト,腸管ガングリア細胞など神経堤由来細胞の発生異常を本態とする稀な疾患である15).臨床的には,中枢神経系の髄鞘形成不全に加え,末梢神経系では脱髄型ニューロパチーを呈し,さらにWaardenburg症候群,Hirschsprung病を合わせた4症候群を合併する.原因遺伝子はSOX10で,常染色体優性遺伝形式をとるが,多くは突然変異による弧発例である16).
臨床症状
多くの症例で出生直後にHirschsprung病に対する外科的治療を要する.神経症状については,重症例では,生直後より末梢・中枢ともに髄鞘形成がほとんど見られず,早期に死亡の転機をとる.中等度の症例では,精神運動発達遅滞と低緊張,約半数で痙性四肢麻痺を呈するとともに,脱髄型ニューロパチーを合併する.軽症例では軽度の運動発達遅滞と脱髄型ニューロパチーを呈する.Waardenburg症候群に関しては虹彩や毛髪,皮膚等の部分的な低色素と感音性難聴を呈する.
検査所見
頭部MRIでは,T2強調画像にて様々な程度で大脳白質のび漫性高信号を認める16).重症例では白質の形成不全による脳幹や小脳,大脳白質の萎縮を認める.一方,軽症例では脳室周囲白質の軽微な高信号を認める.末梢神経伝導速度の低下も様々な程度で認める.
遺伝子診断
ほとんどの症例は,原因遺伝子SOX10のコード領域の点変異によるもので,DNA塩基配列決定法により検出する.多くは患児に新規の変異として起こる.SOX10を含むゲノム領域の欠失の報告もある.
(4)その他,分類不能な疾患
4.おわりに
先天性大脳白質形成不全症の診断は,特異的生化学マーカーがほとんどないため,MRIによる画像診断と遺伝子解析とともに臨床所見に頼らざるを得ない.本稿では,11疾患の分類と概要を明らかにしたが,依然これらの分類に当てはまらないと思われる症例が少なからず存在する.こういった症例が,未知の遺伝子変異による新たな疾患なのか,あるいは既知の遺伝子異常による疾患であるのか,残念ながら現在本邦で可能な遺伝子解析体制でこれを明らかにすることは容易ではない.希少性疾患である先天性大脳白質形成不全症の疾患概念と病態をさらに明らかにしていくためには,1例1例の症例を積み重ねていく以外に方法はない.我々は,本稿にまとめた診断基準が臨床現場で役立つことを期待するとともに,既知疾患の表現型の多様性や新たな疾患の同定により,さらに改訂されていくことを望んでいる.
本稿の診断基準は,平成21年度厚生労働科研費研究報告書に報告された診断基準の内容(一部修正・加筆)に基づいたものである.本研究は,厚生労働科研費難治性疾患克服研究事業「先天性大脳白質形成不全症の診断と治療に向けた研究」により,日本小児神経学会共同研究支援事業の承認を受けて行われている.
文献
- Inoue K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics 2005;6:1-16.
- Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72:750-9.
- 「先天性大脳白質形成不全症の診断と治療に向けた研究」班. 先天性大脳白質形成不全症の診断基準・治療指針. 先天性大脳白質形成不全症の診断と治療に向けた研究 平成21年度総括・分担報告書. 2010;57-75.
- Uhlenberg B, Schuelke M, Ruschendorf F, et al. Mutations in the gene encoding gap junction protein α12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet 2004;75:251-60.
- Orthmann-Murphy JL, Enriquez AD, Abrams CK, Scherer SS. Loss-of-function GJA12/Connexin47 mutations cause Pelizaeus-Merzbacher-like disease. Mol Cell Neurosci 2007;34:629-41.
- van der Knaap MS, Linnankivi T, Paetau A, et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: follow-up and pathology. Neurology 2007;69:166-71.
- van der Knaap MS, Naidu S, Pouwels PJ, et al. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol 2002;23:1466-74.
- Gay CT, Hardies LJ, Rauch RA, et al. Magnetic resonance imaging demonstrates incomplete myelination in 18q- syndrome: evidence for myelin basic protein haploinsufficiency. Am J Med Genet 1997;74:422-31.
- Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet 2004;74:168-75.
- Vaurs-Barriere C, Deville M, Sarret C, et al. Pelizaeus-Merzbacher-Like disease presentation of MCT8 mutated male subjects. Ann Neurol 2009;65:114-8.
- Magen D, Georgopoulos C, Bross P, et al. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet 2008;83:30-42.
- Sasaki M, Takanashi J, Tada H, Sakuma H, Furushima W, Sato N. Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev 2009;31:582-7.
- Zara F, Biancheri R, Bruno C, et al. Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat Genet 2006;38:1111-3.
- Wolf NI, Harting I, Boltshauser E, et al. Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology 2005;64:1461-4.
- Inoue K, Tanabe Y, Lupski J. Myelin deficiencies in both the central and peripheral nervous system associated with a SOX10 mutation. Ann Neurol 1999;46:313-8.
- Inoue K, Khajavi M, Ohyama T, et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet 2004;36:361-9.
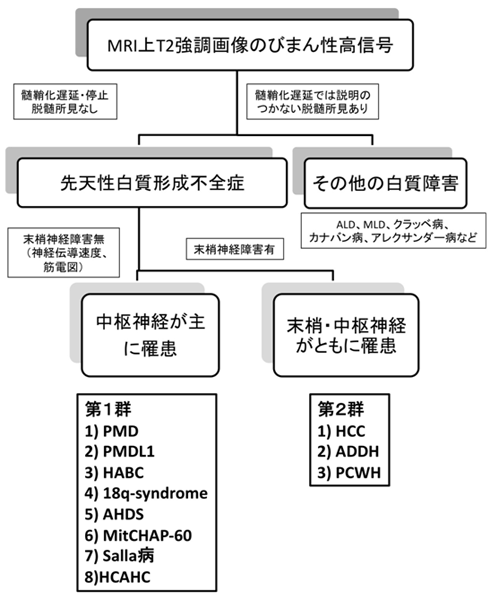
臨床所見からみた先天性大脳白質形成不全症の分類.末梢神経障害の有無により,第1群と第2群に分類される.
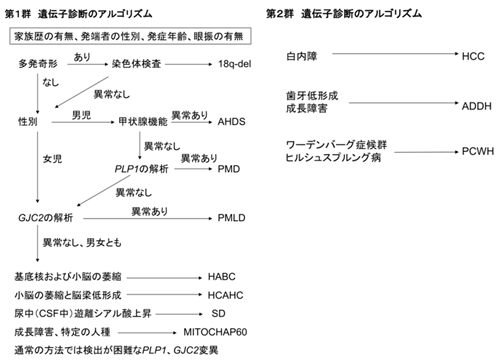
左:第1群疾患の遺伝子診断アルゴリズム.右:第2群疾患の遺伝子診断アルゴリズム.
表1 先天性大脳白質形成不全症の分類
| 疾患名 | 遺伝形式 | 遺伝子座 | 原因遺伝子 |
| 主として中枢神経の先天性髄鞘形成不全を呈する疾患群 | |||
| ペリツェウス・メルツバッハ病 | X劣 | Xq22 | PLP1 |
| ペリツェウス・メルツバッハ様病1 | 常劣 | 1q42.13 | GJC2 |
| 基底核および小脳萎縮を伴う髄鞘形成不全症 | 常優 | 19p13.3 | TUBB4A |
| 18q欠失症候群 | 染色体 | 18q22-qter | MBP |
| アラン・ハーンドン・ダドリ−症候群 | X劣 | Xq13.2 | SLC16A2 |
| Hsp60シャペロン病 | 常劣 | 2q33.1 | HSPD1 |
| サラ病 | 常劣 | 6q13 | SLC17A5 |
| 小脳萎縮と脳梁低形成を伴うび漫性大脳白質形成不全症 | 常劣 | 12q23.3 | POLR3B |
| 中枢神経および末梢神経の先天性髄鞘形成不全を呈する疾患群 | |||
| 先天性白内障を伴う髄鞘形成不全症 | 常劣 | 7p15.3 | FAM126A |
| 失調、歯牙低形成を伴う髄鞘形成不全症 | 常劣 | 10q22.3 | POLR3A |
| 脱髄型末梢神経炎、中枢性髄鞘形成不全症、ワーデンバーグ症候群 | 常優 | 22q13 | SOX10 |
| PLP1, proteolipid protein 1; GJC2, gap junction protein; TUBB 4A, tubulin β-4A; MBP, myelin basic protein; POLR3A, B, two subunits (A and B) of RNA polymerase III; SLC16A2; solute carrier family 16 (monocarboxylic acid transporter), member 2 encoding triiodothyronine, T3, and thyroxine, T4 transporter, monocarboxylate transporter 8 (MCT8); HSPD1, encodes heat shock 60 kD protein, FAM126A, family with sequence similarity 126, member A, DRCTNNB1A; SOX 10, sry-related hmg-box gene 10 | |||









