濃化異骨症
(Pycnodysostosis)
[Synonyms:Pyknodysostosis, Toulouse-Lautrec Syndrome]
Gene Reviews著者: Shannon LeBlanc, MBBS and Ravi Savarirayan, MBBS, MD, FRACP, ARCPA (Hon).
日本語訳者: 佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2020.11.5. 日本語訳最終更新日: 2023.1.7.
原文: Pycnodysostosis
要約
疾患の特徴
濃化異骨症は、四肢短縮型低身長、特徴的顔貌(凸型鼻堤,下顎角の開大を伴う小下顎症)、骨脆弱性亢進を伴う骨硬化症、末節骨の先端骨溶解症、頭蓋縫合閉鎖遅延、鎖骨異形成を特徴とする。罹患者の示す顔貌の特徴は、顔面骨に生じる進行性の先端骨溶解に伴うと思われる変化により、年齢とともにより顕著になっていくものの、ふつうは幼少期にあっても、小下顎と凸型鼻堤を中心に、その特徴は十分認知しうる程度に存在する。その他の症候としては、歯や爪の異常がある。知能はふつう正常であるが、一部、軽度の精神運動障害を有する例も報告されている。
診断・検査
発端者における濃化異骨症の診断は、特徴的な臨床症候とX線写真症候、ないしは分子遺伝学的検査でCTSKの両アレルの病的バリアントが同定されることにより確定する。
臨床的マネジメント
症候に対する治療:
成長ホルモン治療、必要に応じて行う生活環境・活動環境の改善、骨折や脊柱側彎に対する整形外科的治療、口蓋裂・頭蓋縫合早期癒合・上下顎骨低形成に関し必要に応じて行う頭蓋顔面外科的・神経外科的処置、閉塞性睡眠時無呼吸に対する呼吸・睡眠医学の専門家による管理、予定手術に先立って行う麻酔科医との協議、歯の異常に対する歯科的・矯正歯科的治療、視覚の問題に関する眼科医による標準治療といったものがある。
定期的追跡評価:
脊柱側彎、非対称、骨折頻度、体重・栄養、心理面に関して行う年に1度の身体的診査に加え、睡眠ポリグラフを2年に1度、専門の歯科医、眼科医による診査を年に1度行う。
避けるべき薬剤/環境:
全身麻酔を要する場合、麻酔日程の決定に先立って、挿管困難の可能性について考えておく必要がある。
妊娠に関する管理:
狭骨盤の罹患者については、帝王切開による分娩を検討する必要がある。ただ、この点については、症例ごとに、骨系統疾患に明るい産科医と麻酔科医による評価が必要である。
遺伝カウンセリング
濃化異骨症は、常染色体潜性の遺伝形式をとる。両親ともCTSKの病的バリアントをヘテロで有していることがわかっている場合、罹患者の同胞は、受胎の段階で罹患者である可能性が25%、無症状の保因者である可能性が50%、罹患者でも保因者でもない可能性が25%である。家系内に存在するCTSKの病的バリアントが同定されている場合は、リスクを有する血族に対する保因者確認の検査、高リスクの妊娠に備えた出生前診断、着床前遺伝学的検査が可能となる。
診断
濃化異骨症の公式な診断基準は確立されていないものの、X線写真でみられる先端骨溶解症、骨硬化症、正常な下顎角の消失といった症候が、ほぼ疾患特異的と言えるものである。
本疾患を示唆する所見
以下のような臨床所見、X線写真所見、検査所見を有する例については、濃化異骨症を疑う必要がある。
臨床所見
- 四肢短縮型低身長が全例でみられる(出生前の発症が30%近く)。
- 短指趾
- 頭蓋顔面所見
- 前額部の突出
- 大泉門の閉鎖遅延
- 凸型鼻堤を伴う目立つ鼻
- 上顎骨・下顎骨の低形成に起因する中顔面の後退と小下顎
- 喘鳴,喉頭軟化症,閉塞性睡眠時無呼吸
- 青みがかった強膜を伴う突出した眼
- 高口蓋/口蓋の溝形成
- 歯の異常(例えば、乳歯・永久歯の萌出遅延,乳歯の晩期残存に伴う乳歯・永久歯の併存,部分性無歯症)
- 爪の奇形(例えば、異形成,溝形成,平坦化)
X線写真所見
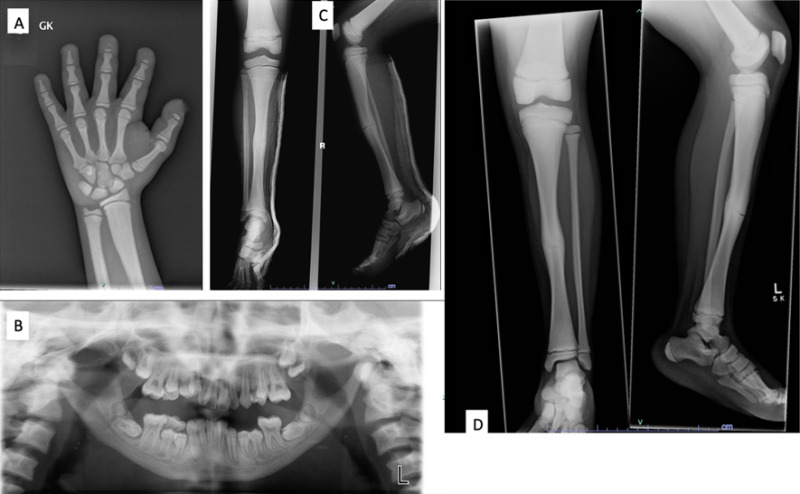
図1:濃化異骨症のX線写真像
A.12歳女性の手根骨X線写真。末節骨の顕著な先端骨溶解と、全体的な骨密度の上昇がみられる。
B.同一罹患者の混合歯列期のパノラマX線写真。下顎角がみられず、下顎骨は低形成と骨硬化の様相を呈する。
C.10歳時の脛骨と腓骨のX線写真。瀰漫性の骨硬化と、骨幹中央部の横骨折がみられる。
D.骨膜性骨形成を伴いつつ、骨折は3ヵ月後もはっきり見える形で残っている。
- 長管骨を中心とした全身性、進行性の骨硬化
- 末節骨の先端骨溶解
- 乳突蜂巣の無含気
- 頭蓋縫合癒合遅延
- 正常な下顎角が形成されないことに起因する下顎角の開大
- 骨折の頻発
- 鎖骨異形成,鎖骨の先天性偽関節
臨床検査所見
- 血清カルシウム・リン・ビタミンD・アルカリホスファターゼはいずれも正常。
- 成長ホルモン分泌低下。
- インスリン様成長因子1(IGF-1)が低値。
- その他の下垂体ホルモンに異常なし。
家族歴
常染色体潜性遺伝に合致した家族歴(例えば、罹患同胞の存在,両親の血族結婚)がみられる。ただし、家族歴がなくても、濃化異骨症の診断が除外されるわけではない。
診断の確定
発端者における濃化異骨症の診断は、特徴的な臨床症候とX線写真症候がみられること、もしくは分子遺伝学的検査でCTSKの両アレルの病的バリアントが同定されることにより確定する(表1参照)。
注:CTSKの両アレルに意義不明のバリアントが検出された場合(もしくは、CTSKの一方のアレルに病的バリアント、他方のアレルに意義不明のバリアントが検出された場合)、それは濃化異骨症の診断を確定させるものでも排除するものでもない。
分子遺伝学的検査としては、表現型に合わせて、遺伝子標的型検査(単一遺伝子検査,マルチ遺伝子パネル)と網羅的ゲノム検査(エクソームシーケンシング,ゲノムシーケンシング)とを組み合わせて用いることが考えられる。
遺伝子標的型検査の場合、臨床医のほうで、どの遺伝子の関与が疑われるか目星をつけておく必要があるが、ゲノム検査の場合、その必要はない。「本疾患を示唆する所見」に書かれた特徴的症候を有する例については遺伝子標的型検査(「方法1」参照)で診断がつく可能性が高く、一方、表現型からは骨硬化や低身長を伴うその他数々の遺伝性疾患との鑑別が難しい例については、ゲノム検査(「方法2」参照)で診断がつく可能性が高い。
方法1
表現型やX線写真所見から濃化異骨症の診断が示唆される場合、分子遺伝学的検査としては、単一遺伝子検査、もしくはマルチ遺伝子パネルの使用が考えられる。
- 単一遺伝子検査 遺伝子内の小欠失/挿入、ならびにミスセンス・ナンセンス・スプライス部位バリアントを検出する目的で、最初にCTSKの配列解析を行う。この手法では、通常、エクソン単位あるいは遺伝子全体の欠失/重複は検出されない。
- マルチ遺伝子パネル 意義不明のバリアントや、現況の表現型と関係のない遺伝子の病的バリアントの検出を抑えつつ、疾患の遺伝的原因の特定に至る可能性が最も高いと思われるのは、CTSKその他の関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネルである。
注:(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、今このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。遺伝子検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
方法2
表現型からだけでは、骨硬化や低身長を呈するその他数々の遺伝性疾患と区別しづらいといった場合であれば、網羅的ゲノム検査(この場合、臨床医の側でどの遺伝子の関与が疑われるかを検討する必要はない)が選択肢となる。エクソームシーケンシングが最も広く用いられるが、ゲノムシーケンシングを選択することも可能である。
網羅的ゲノム検査の基礎的情報についてはここをクリック。ゲノム検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
表1:濃化異骨症で用いられる分子遺伝学的検査
| 遺伝子1 | 手法 | その手法で病的バリアント2が検出される割合 |
|---|---|---|
| CTSK | 配列解析3 | 100%近く4 |
| 遺伝子標的型欠失/重複解析5 | 1例の報告あり6 |
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- この遺伝子で検出されているバリアントの情報については、「分子遺伝学」の項を参照のこと。
- 配列解析を行うことで、benign、likely benign、意義不明、likely pathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、遺伝子内の小さな欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。
配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。 - 論文の形で公表され閲覧可能な全症例、ClinVar[Landrumら2014]、ヒト遺伝子変異データベース(HGMD)[Stensonら2017]の購読ベースの専門家向けデータを合わせた、おおむね35種の病的バリアントについてレビューを行ったもの。
- 遺伝子標的型欠失/重複解析では、遺伝子内の欠失や重複が検出される。具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失/重複の検出を目的に設計された遺伝子標的型マイクロアレイなど、さまざまなものがある。
- イントロン7への301bpのAlu配列の挿入。これにより、新たなスプライス受容部位が形成される可能性がある[Armanら2014]。
臨床的特徴
臨床像
濃化異骨症は、低身長、特徴的顔貌(下顎角の開大を伴う小下顎,凸型鼻堤)、骨脆弱性亢進を伴う骨硬化、末節骨の先端骨溶解、頭蓋縫合閉鎖遅延、鎖骨異形成を特徴とする。
罹患者の顔貌の特徴は、年齢とともにより顕著になる。これは、顔面骨に生じる進行性の先端骨溶解に起因するものと考えられるものの、小下顎や凸型鼻堤を中心に、その特徴は、幼少期においても十分認知できる程度に存在することが多い[Turan 2014]。
先に公表された広範なレビュー報告[Xueら2011]によると、CTSKの病的バリアントのホモ接合ないし複合ヘテロ接合が確認された例は、59家系、159例存在する。これとは別に、最近、17家系、27例が報告されており、うち14家系で分子レベルのデータが明らかになっている[Bizaouiら2019]。濃化異骨症の表現型の特徴として述べる以下の記述は、これらの報告を基にしたものである。
表2:濃化異骨症:代表的症候の出現頻度
| 症候 | その症候を有する例の割合 | |
|---|---|---|
| 臨床症候 | 四肢短縮,低身長 | 100%近く |
| 子宮内成長抑制 | 30%近く | |
| 短指趾 | 90%超 | |
| 前額部の突出 | 80%超 | |
| 大泉門開存 | 80% | |
| 凸型鼻堤 | 70%近く | |
| 小下顎 | 70%超 | |
| 中顔面の後退 | 60% | |
| 眼球突出 | 60% | |
| 青みがかった強膜 | 30%-40% | |
| 睡眠時無呼吸 | 65%超 | |
| 骨折頻度の上昇 | 70%近く | |
| 爪の奇形 | 50%超 | |
| 歯の異常 | 30%-40% | |
| X線写真症候 | 骨硬化 | 100%近く |
| 末節骨の先端骨溶解 | 90%超 | |
| 乳突蜂巣の無含気 | 80% | |
| 頭蓋縫合の閉鎖遅延 | 67% | |
| 下顎角の開大 | 65% | |
| 鎖骨異形成 | 25% | |
成長障害/低身長
濃化異骨症罹患者のほぼ100%で、低身長が報告されている。
通常、罹患者は幼児期までに成長速度の低下が始まり、低身長に至るが、30%については、子宮内の段階ですでに成長障害を示すと報告されている。
四肢は、近位肢・中間肢・遠位肢のそれぞれが短小で、多くの場合、体幹に対して不均衡型の短小を示す。
記録にある成人期の身長は、通常、男性については150cm未満(平均すると平均身長の-2.9SD)、女性については130-134cm(平均すると平均身長の-4.1SD)である[Bizaouiら2019]。
約50%に成長ホルモン分泌不全がみられるが、IGF-1値については、ほぼ全例が低値を示す。成長ホルモンの投与によりIGF-1値が良好に上昇し、成人期の身長や骨格の均整は正常に近いところに落ち着く[Rothenbühlerら2010]。
成長ホルモン分泌不全を有する例の多くにおいては、頭部の画像で下垂体低形成が併せて確認される。ただ、他の下垂体ホルモンの異常や、思春期発達の異常等はみられない[Turanら2014]。
これまでに、153cm(-1.9SD)のメキシコ人成人男性、150cm(-0.6SD)のメキシコ人成人女性、正常な身長(137cm;-0.9SD)を有する11歳の中国人男児といった、通常より高い身長を有する3例(2例は臨床診断、1例は分子診断)が報告されている[Zhengら2013,Valdes-Floresら2014]。
頭蓋顔面の外観
特徴的顔貌(上顎低形成に起因する中顔面の後退,下顎角の開大を伴う小下顎)は年齢とともにより顕著になっていくが、乳児期においても、大きな大泉門と小泉門、前額部や頭頂部の突出を伴う頭蓋縫合の離開といった形で、多くの場合、認知可能である[Appelman-Dijkstra & Papapoulos 2016]。よくみられるそれ以外の顔面症候に凸型鼻堤がある。それよりは少ないものの、青みがかった強膜を伴う眼球突出や、正中の溝形成を伴う高口蓋ないし口蓋裂がみられることもある[Bizaouiら2019]。Otaifyら[2018]は、8例について調査を行い、口蓋の溝という形で見えるものは、口蓋の幅径が小さく彎曲の程度も少ないこと、ならびに上顎骨鼻稜が落ち込んで目立つ形の口蓋縫線を形成したことに起因するものであるとしている。
骨格
低身長の次に多くみられる症候は、骨密度の上昇(骨硬化)で、これは骨格全体にわたって生じ、かつ進行性である。髄管は狭いながらも存在し、造血機能も維持されていることがわかっている。
短指趾と指趾末節骨の進行性先端骨溶解を伴う短い手足が報告例の90%超でみられる。
中手骨・中足骨の短小に関する報告はみられない。
画像でみられるその他の症候としては、乳突蜂巣の無含気(80%)と、頭蓋冠の縫合の癒合遅延(67%)がある。鎖骨は肩峰端の先端骨溶解を伴う異形成状態を示すことがある(25%)。
これらより少ない症候としては、ウォルム骨(18%)、軽度の脊柱側彎(12%)、下肢長の不均衡(8%)、脊椎分離症、脊椎すべり症、狭い腸骨などがある。冠状縫合早期癒合の報告が4例みられる[Bertolaら2010,Caracasら2012,Bizaouiら2019]。慢性痛の報告が濃化異骨症成人の60%に及ぶ。その発症時期は、20歳代であることが多い[Bizaouiら2019]。
骨の脆弱性
濃化異骨症罹患者においては、年平均0.2件と、骨折率の上昇がみられ、初回骨折年齢は10歳前後である[Bizaouiら2019]。報告された範囲で最も低年齢の骨折例は生後10ヵ月である。この例については、同じ疾患で死亡に至った2同胞が存在することから、重度型の表現型ないし遺伝型であったと思われる。ただ、分子レベルの検査は行われていない[Caracasら2012]。
骨折の治癒はしばしば遅延し、リモデリングも不完全に終わる。髄管の狭窄のため、外科的固定は困難なことが多く、それに加えて、骨硬化のため術中の医原性骨折リスクが高まることになる[Grewalら2019]。骨の脆弱性に関しては、有効な薬物療法は現在に至るまで確立されていない。濃化異骨症は、背後に破骨細胞の機能異常の問題があるため、ビスホスホネート治療は禁忌である。
耳鼻咽喉
喘鳴や喉頭軟化症(20%)は、症候としてそれほど珍しくはなく、これがあることで早期に濃化異骨症が疑われるきっかけにもなる。睡眠時無呼吸(OSA)の報告もしばしばみられ(60%超)、濃化異骨症児ではこれが特に重度な場合がある。OSAを伴う例の48%で、5歳から10歳の間に非侵襲的な換気療法が必要になる[Testaniら2014,Bizaouiら2019]。軽度の伝音性難聴を呈する例は、罹患者の50%に及ぶ[Bizaouiら2019]。
歯
歯の異常としては、乳歯・永久歯の萌出遅延、乳歯の晩期残存(その結果、後継永久歯と併存する形になる)、部分性無歯症、咬合異常、エナメル質形成不全、齲蝕の増加などがある[Turan 2014,Khojaら2015,Otaifyら2018]。
爪
爪の平坦化、溝形成、異形成がしばしばみられる。指の短小化と先端骨溶解に伴う二次性の変化として、指の背側の皮膚に皺が寄ることがある。
神経
脳奇形を伴う場合を除き、ふつう濃化異骨症罹患者の知能は正常である。軽度の精神運動障害が罹患者の30%にみられると報告されている[Bizaouiら2019]。稀に報告のある神経学的異常に、ChiariⅠ型奇形(1例)、脳の脱髄(3例)、錐体路症候群(1例)がある[Solimanら2001,Stark & Savarirayan 2009,Bizaouiら2019]。
眼
これまでに屈折障害、斜視等の眼の異常が報告されている。頭蓋内圧亢進と乳頭浮腫の結果、重度の視力低下をきたした1例が報告されている[Bizaouiら2019]。
肥満
濃化異骨症の典型的症候として、これまでに肥満が報告されているわけではない。しかしながら、27人のコホート中の26%に体重過多がみられている[Bizaouiら2019]。
予後
濃化異骨症罹患者の寿命は、ふつう正常である。
その他
それほど多くはない報告として、関節弛緩、胸の変形(狭い胸郭,脊柱後彎,脊柱前彎)、肝脾腫などがある。骨盤偏腎ならびに原因不明の汎血球減少症が、これまでに各1例報告されている。
遺伝型-表現型相関
rCTSKの遺伝型-表現型相関としてこれまでに確認されているものはない。
命名法について
濃化異骨症(pycnodysostosis:ギリシア語でpycno=密,dys=欠陥のある,osteon=骨)は、1962年、Maroteaux and Lamyによって最初の報告がなされた。そのため、Maroteaux-Lamy症候群の名でも知られる[Xueら2011,Bizaouiら2019](この名称は、もともと、濃化異骨症とは全く別の、ARSBの病的バリアントによって生じるムコ多糖症Ⅳ型を指す病名として用いられていた)。
濃化異骨症はまた、時として、フランス人画家、Henri de Toulouse-Lautrec(1864-1901)に因んで、「Toulouse-Lautrec症候群」と呼ばれることもある。低身長、親の血族結婚、顔の異形、骨折の頻発、大きな大泉門といった濃化異骨症に合致する表現型の特徴をもとに、Lautrecはおそらくこの疾患であっただろうと回顧的に考えられている[Turan 2014](図2参照)。

図2:濃化異骨症罹患者と考えられる画家Henri de Toulouse-Lautrecの1898年の肖像画,Edouard Vuillard (1868-1940)作
発生頻度
医学文献において、これまでに約200例が報告されている。濃化異骨症の発生頻度は100万人あたり1-1.7人と推定されている。
遺伝学的に関連のある疾患(同一アレル疾患)
CTSKの生殖細胞系列の病的バリアントに関連して生じるものとしては、このGeneReviewで述べたもの以外の表現型は知られていない。
鑑別診断
ある種の大理石骨病では、早期の造血幹細胞移植が治療上の選択肢となりうるが、濃化異骨症の場合は骨髄不全を示すことがほとんどないため、これが有益な選択肢となることはない[Bizaouiら2019]。そのため、濃化異骨症については、大理石骨病の特徴を有する他の原発性骨硬化性疾患(表3参照)との鑑別がきわめて重要である。
表3:濃化異骨症との鑑別に係わってくる大理石骨病を伴う疾患群
| 鑑別を要する疾患でみられる濃化異骨症と重なる症候 | 遺伝子 | 鑑別を要する疾患 | 遺伝形式 | 鑑別を要する疾患でみられるものの、濃化異骨症ではみられない症候 |
|---|---|---|---|---|
| 骨硬化症,さまざまな重症度の瀰漫性、局在性硬化症,骨幹端におけるモデリング障害, 骨髄炎,病的骨折, 歯の萌出障害 |
CA2 | 腎細管性アシドーシスを伴う大理石骨病 (OMIM 259730) |
AR | 骨髄障害は稀。 脳神経圧迫,発達遅滞,頭蓋内石灰化,腎尿細管性アシドーシス |
| CLCN7 SNX10 TCIRG1 |
大理石骨病,重症新生児型/乳児型 (「CLCN7関連大理石骨病」のGeneReviewを参照) |
AR | 脳神経圧迫(Ⅱ,Ⅶ,Ⅷ),髄外造血,水頭症,低カルシウム血症,汎血球減少症 | |
| CLCN7 PLEKHM1 TNFSF11 |
大理石骨病,中間型1 (「CLCN7関連大理石骨病」のGeneReviewを参照) |
AR | 貧血,髄外造血,時に視神経圧迫 | |
| CLCN7 | 大理石骨病,遅発型2型 | AD | 中等度の造血障害,脳神経の圧迫 | |
| FERMT3 | 大理石骨病,白血球接着不全を伴う中等症型 (OMIM 612840) |
AR | 好中球の内皮細胞への接着不全,肝脾腫,白血球増多,粘膜出血 | |
| IKBKG | 外胚葉異形成と免疫不全を伴う大理石骨病 (OMIM 300291) |
XL | 低汗性外胚葉異形成,免疫不全(重篤な感染を惹起),リンパ浮腫 | |
| OSTM1 | 大理石骨病,乳児型,神経系の罹患を伴う (OMIM 259720) |
AR | 脳神経圧迫(Ⅱ,Ⅶ,Ⅷ),髄外造血,水頭症,低カルシウム血症,汎血球減少症,網膜萎縮を含む原発性神経変性 | |
| TNFRSF11A | 大理石骨病,乳児型,免疫グロブリン欠乏を伴う破骨細胞減少型 (OMIM 612301) |
AR | 貧血,肝脾腫,低γグロブリン血症,血小板減少症 | |
| 骨硬化症,低身長, 病的骨折 |
CSF1R TNFRSF11A SLC29A3 |
異骨性骨異形成症 (OMIM 618476)2 |
AR | 脳の異常,進行性の神経学的悪化(CSF1Rに特異的),皮膚の高色素沈着パッチ,扁平椎,管状骨の骨幹端下拡大部の放射線透過性 |
| 主に付属骨の骨幹端と骨端縁、軸骨格を構成する骨の骨幹端相当部に限局して現れる骨硬化 | LRRK1 | 骨硬化性骨幹端異形成症3 | AR | 発達遅滞,ピリジノリン・デオキシピリジノリンの尿中排泄の上昇,血清ALP・AST・CK値の上昇,癲癇発作 |
| 先端骨溶解,関節弛緩,低身長,頭蓋骨変形 | NOTCH2 | Hajdu-Cheney症候群 (OMIM 102500) |
AD | 軽度の知的障害(ごく一部),骨粗鬆症 |
| 鎖骨異形成,大泉門閉鎖遅延,歯の萌出遅延,高口蓋,低身長 | RUNX2 | 鎖骨頭蓋異形成スペクトラム障害 | AD | 骨盤・恥骨の形態異常,鎖骨欠損,胸郭変形 |
AD=常染色体顕性,AR=常染色体潜性,XL=X連鎖性
- Liら[2019]
- Campeauら[2012],Xueら[2019]
- Iidaら[2016]
続発性に骨硬化症を生じさせる要因
濃化異骨症、ならびにその他の破骨細胞の機能障害に起因して生じる原発性骨硬化性疾患は、続発性に骨硬化を生じさせる数々の要因と区別する必要がある。検討を要するそうした要因としては、フッ素症、ベリリウム・鉛・ビスマスの中毒、骨髄線維症、Paget病骨硬化型(OMIM PS167250)、悪性腫瘍(リンパ腫,造骨性癌転移)などがある[Stark & Savarirayan 2009]。
臨床的マネジメント
濃化異骨症の治療やサーベイランスに関するガイドライン、あるいは、濃化異骨症で発生する骨折への最適な対処法・外科的介入法に関する標準ガイドラインといったものは公表されていない。管理にあたっては、集学的治療を行うこと、ならびに、必要な場合は熟慮を重ねた上で外科的介入を行うといったことを重視しながら進める必要がある。
最初の診断に続いて行う評価
濃化異骨症と診断された罹患者については、疾患の範囲やニーズを把握するため、診断に至る過程ですでに実施済ということでなければ、表4にまとめたような評価を行うことが推奨される。
表4:濃化異骨症罹患者において最初の診断後に行うことが推奨される評価
| 系/懸念事項 | 評価項目 | コメント |
|---|---|---|
| 体型 |
|
体重管理に関して、必要なら栄養士への紹介を検討。 |
| 筋骨格系 | 脊椎の側面撮影を含むX線写真による徹底した骨格の検査 | |
| 頭蓋のCTを検討する | 頭蓋縫合早期癒合症の臨床的懸念がある場合。 | |
| 整形外科への受診 | 可能であれば、骨系統疾患に明るい専門医による評価 | |
| 耳鼻咽喉系 |
|
|
| 呼吸器系 | 睡眠ポリグラフ | 全罹患者について、実施可能な範囲でできるだけ早期に行う。 |
| 歯 | 治療開始時点の記録としての歯科的評価 | |
| 神経系 | MRIを検討する。 | Chiari奇形に関する神経症状ないし懸念がある場合。 |
| 眼 | 治療開始時点の記録としての眼科的診査 | |
| 遺伝カウンセリング | 遺伝の専門職1の手で行う。 | 医学的・個人的決断に資するよう、罹患者とその家族に対し、濃化異骨症の本質・遺伝形式・そのもつ意味について情報提供を行う。 |
| 家族への支援/情報資源 | 以下の諸点に関し評価を行う。
|
- 臨床遺伝医,認定遺伝カウンセラー,認定上級遺伝看護師をいう。
表5:濃化異骨症罹患者の症候に対する治療
| 症候/懸念事項 | 治療 | 考慮事項/その他 |
|---|---|---|
| 成長ホルモン分泌不全/低身長 |
|
|
| 骨折 |
|
|
| 脊柱側彎 | 整形外科医による管理 | |
| 頭蓋顔面 | 口蓋裂・頭蓋縫合早期癒合・上下顎の低形成に関し、必要に応じ、頭蓋顔面外科的/脳神経外科的管理 | 上下顎の骨延長術も選択肢となりうる。 |
| 閉塞性睡眠時無呼吸 |
|
気道狭窄に起因する鼻閉に注意。 |
| 麻酔に関する必要事項 | 予定手術の際は常に事前に経験豊富な麻酔科医と相談を行う。 | 挿管困難のリスクがありうる。 |
| 歯 |
|
骨密度上昇に起因して、抜歯後骨髄炎のリスクが高まる |
| 視覚に関する懸念 | 眼科医による標準管理 |
定期的追跡評価
表6:濃化異骨症罹患者で推奨される定期的追跡評価
| 系/懸念事項 | 評価項目 | 実施頻度 |
|---|---|---|
| 全身の健康 | 身体の診査 | 年に1度、もしくは指示通りに |
| 筋骨格系 |
|
年に1度 |
| 呼吸器系 | 睡眠ポリグラフ | 2年に1度 |
| 歯 | 専門歯科医による評価 | 年に1度 |
| 視覚 | 眼科的診査 | |
| 肥満 | 体重の評価。 これに栄養士のチェックを加えることあり。 |
年に1度、もしくは指示通りに |
| 心理 | 病歴聴取や身体の診査を行う際に、心理面に何らかの問題がないか、注意しながら進める。 |
Bizaouiら[2019]
避けるべき薬剤/環境
全身麻酔を要する例については、麻酔計画に先立って、挿管困難の可能性について検討しておく必要がある。
濃化異骨症は、背景に破骨細胞の機能異常が存在するため、ビスホスホネート治療は禁忌である。
リスクを有する血縁者の評価
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
妊娠に関する管理
狭骨盤の罹患者については、帝王切開による分娩を検討する必要がある。ただし、その場合でも、骨系統疾患に明るい産科医や麻酔科医による評価が必須である[Savarirayanら2018]。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「Clinical Trials.gov」、ならびにヨーロッパの「EU Clinical Trials Register」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
濃化異骨症は、常染色体潜性の遺伝形式をとる。
家族構成員のリスク
発端者の両親
- 罹患児の両親は、絶対ヘテロ接合者である(すなわち、族歴に照らして、CTSKの病的バリアントを1つ保有する保因者と推定される)。
- 発端者の分子遺伝学的結果が判明している場合には、両親に対しても分子遺伝学的検査を行って、両親ともCTSKの病的バリアントのヘテロ接合者であることを確認するとともに、再発リスク評価を信頼性のあるものにしていく必要がある。(常染色体潜性遺伝疾患においては、低いながらも一定の割合で新生のバリアントが発生することが知られている[Jónssonら2017]。)
- ヘテロ接合者(保因者)は無症状であり、本疾患の発症リスクは有しない。
発端者の同胞
- 両親とも、CTSKの病的バリアントをヘテロで有していることが判明している場合は、受胎の段階で同胞が罹患者である可能性が25%、無症状の保因者である可能性が50%、罹患者でも保因者でもない可能性が25%となる。
- CTSKの病的バリアントを両アレル性に継承した同胞間にあっても、その臨床症候には家系内で幅がみられる可能性がある。
- ヘテロ接合者(保因者)は無症状であり、本疾患の発症リスクは有しない。
発端者の子
濃化異骨症罹患者の子は、CTSKの病的バリアントに関し、絶対ヘテロ接合者(絶対保因者)となる。
他の家族構成員
発端者の両親の同胞は、CTSKの病的バリアントの保因者であることに関し、50%のリスクを有している。
保因者の特定
リスクを有する血族に対して保因者の検査を行うにあたっては、先に家系内に存在するCTSKの病的バリアントを同定しておく必要がある。
関連する遺伝カウンセリング上の諸事項
家族計画
- 遺伝的リスクの確定、保因者であることの明確化、出生前/着床前遺伝学的検査を受けるかどうかの話し合いといったことに最も適しているのは、妊娠前の時期である。
- 罹患者、保因者、ならびに保因者である可能性のある若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法であるとか、遺伝子・アレルバリアント・疾患等に対するわれわれの理解が、将来はより進歩していくことが予想される。そのため、分子診断の確定していない(すなわち、原因となった遺伝子の変化が未解明の)発端者のDNAについては、保存することを検討すべきである。
出生前検査ならびに着床前遺伝学的検査
家系内に存在するCTSKの病的バリアントが同定されている場合は、高リスクの妊娠に備えた出生前検査や、着床前遺伝学的検査を行うことが可能である。
出生前検査の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。
現在、多くの医療機関では、出生前検査を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- Human Growth Foundation (HGF)
- MAGIC Foundation
997 Glen Cove Avenue
Suite 5
Glen Head NY 11545
Phone: 800-451-6434 (toll-free)
Fax: 516-671-4055
Email: hgf1@hgfound.org
www.hgfound.org
4200 Cantera Drive #106
Warrenville IL 60555
Phone: 800-362-4423 (Toll-free Parent Help Line); 630-836-8200
Fax: 630-836-8181
Email: contactus@magicfoundation.org
www.magicfoundation.org
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:濃化異骨症:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specificデータベース | HGMG | ClinVar |
|---|---|---|---|---|---|
| CTSK | 1q21.3 | カテプシンK | CTSK database | CTSK | CTSK |
データは、以下の標準資料から作成したものである。
遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。
リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:濃化異骨症関連のOMIM>エントリー(閲覧はすべてOMIMへ)
| 265800 | PYCNODYSOSTOSIS |
| 601105 | CATHEPSIN K; CTSK |
分子レベルの病原
CTSKはカテプシンKをコードしている。カテプシンKは、骨のリモデリングに関与するリソソーム内システインプロテアーゼで、破骨細胞内において顕著な発現を示す。マクロファージ内、ならびに骨髄由来樹状細胞内においても検出されるが、脾T細胞内にはほとんどみられない。濃化異骨症においては、破骨細胞の数は正常で、波状縁や明帯といった構造にも変化はみられないものの、個々の破骨細胞周囲の脱灰骨の領域が広くなっている。
コラーゲン骨基質の溶解は、マトリックスメタロプロテアーゼとリソソームカテプシンという2群の酵素によって進んでいく。中でもカテプシンKは、鍵となる重要酵素と位置づけられている。カテプシンKは、前酵素として合成された後、リソソームに運ばれ、そこで切断処理を受けて活性型酵素としてのカテプシンKが完成する。カテプシンKは、低pH下で、骨基質タンパク質であるⅠ型・Ⅱ型コラーゲン、オステオポンチン、オステオネクチンの分解に関与する[Stark & Savarirayan 2009,Turan 2014,Appelman-Dijkstra & Papapoulos 2016]。濃化異骨症においては、この分解が低下し、結果として骨密度の上昇が生じる。
疾患発症のメカニズム
機能喪失型である。
CTSK特異的な検査技術に関する考察
- 変異のホットスポットとなっている残基は、Arg241とAla277である[Xueら2011,Turan 2014,Bizaouiら2019]。
- これまでに約60種のCTSKの病的バリアントが報告されている。コードしている領域の内訳は、成熟領域が69%、プロ領域が24%、プレ領域(シグナル配列)が6%である。
- プライマーの選択をよく考えて行うことで、イントロン7の挿入の検出が容易に行えるようになる(表1参照)。
更新履歴:
-
Gene Reviews著者: Shannon LeBlanc, MBBS and Ravi Savarirayan, MBBS, MD, FRACP, ARCPA (Hon).
日本語訳者: 佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2020.11.5. 日本語訳最終更新日: 2023.1.7.[in present]
原文: Pycnodysostosis
![]()