Nicolaides-Baraitser症候群
(Nicolaides-Baraitser Syndrome)
[Synonyms:NCBRS]
Gene Reviews著者: Omar Abdul-Rahman, MD
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2015.10.15. 日本語訳最終更新日: 2022.12.3.
原文 Nicolaides-Baraitser Syndrome
要約
疾患の特徴
Nicolaides-Baraitser症候群は、疎な頭髪、皮下脂肪が少ないことに起因する指節間関節や末節骨の膨隆、特徴的な粗野な顔、小頭症、癲癇発作、発達遅滞/知的障害を特徴とする疾患である。
癲癇発作にはさまざまな種類のものがみられ、管理が難しい場合がある。
発達遅滞/知的障害(ID)は、半数近くが重度、3分の1が中等度、残りが軽度である。
3分の1近くについては、言語発達が全く生じない。
診断・検査
発端者におけるNCBRSの診断は、これを示唆する臨床症候がみられることに加え、分子遺伝学的検査でSMARCA2の病的バリアントのヘテロ接合が同定されることで確立する。
臨床的マネジメント
症候に対する治療:
癲癇発作に対する神経内科医ないし癲癇専門医の管理下での抗痙攣薬(ASM)処方、作業療法・理学療法・言語治療、屈折異常や難聴に対する通法に従った管理を行う。
定期的追跡評価:
癲癇発作の評価や管理に関して、神経内科医による少なくとも年に1度の評価、発達の進行状況や治療的・教育的介入の如何に関する発達小児科医による年に1度の評価、眼科的異常・聴覚異常に関する定期的追跡評価などが必要である。
遺伝カウンセリング
NCBRSは、常染色体顕性の遺伝形式をとる。
現在までに報告されている罹患者は、すべて、新生のSMARCA2の病的バリアントに起因してNCBRSが生じた例である。
罹患同胞の報告はみられないが、片親が生殖細胞系列モザイクである可能性が考えられるため、同胞再現率については一般集団より高くなるものと推定される。
家系内に存在するSMARCA2の病的バリアントが同定されている場合は、高リスクの妊娠となることが理論上考えられることから、出生前検査を行うことが可能となる。
診断
Nicolaides-Baraitser症候群(NCBRS)の臨床的診断基準に関する合意は、現在のところ確立されていない。
本疾患を示唆する所見
以下のような臨床症候やX線症候を有する例については、Nicolaides-Baraitser症候群(NCBRS)を疑う必要がある。
臨床症候(出現頻度の高い順)
- 発達遅滞/知的障害 重度が50%、中等度が30%、軽度が20%。
- 疎な頭髪
- 皮下脂肪の少なさに起因する指節間関節や末節骨の膨隆(図1,図2を参照)
- 特徴的顔貌(前向き鼻孔,長い人中,大きな口,薄い上赤唇,厚い下赤唇) 新生児期、乳幼児期には、顔面の症候は軽微にとどまることがあるものの、経時的に顔の粗野さや皮膚の皺が顕著に現れるようになる(図3参照)。
- 小頭症
- 癲癇発作

図1:指の脂肪沈着が少ないことに起因する指節間関節の膨隆
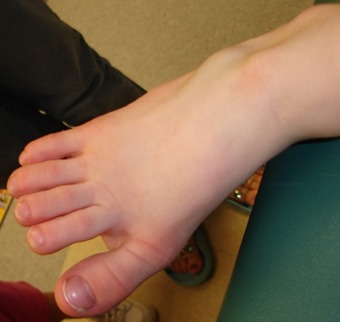
図2 指節間関節の中等度の膨隆
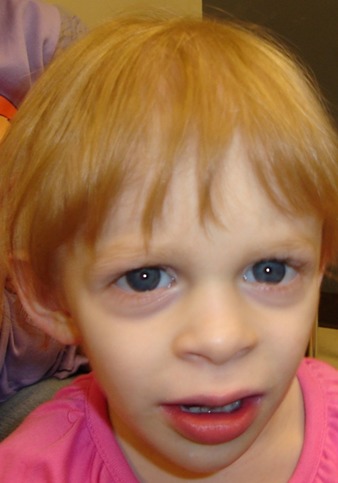
図3 :疎な頭髪、薄い上赤唇、厚い下赤唇を伴う粗野な顔
X線症候
- 手については、円錐形骨端、末広がりの指節骨骨幹端、指節骨・中手骨(中足骨)の短小化(特に、第4・第5指趾列)がみられることがある。
- 骨年齢にはばらつきがみられ、遅延を示す16人の子ども、亢進を示す2人の子どもが報告されている。
- 扁平椎、扁平椎間板、小さい骨盤、恥骨低形成、小さい大腿骨頭、短い大腿骨頸部といったものが報告されている。
診断の確定
発端者におけるNicolaides-Baraitser症候群の診断は、これを示唆する臨床症候がみられることに加え、分子遺伝学的検査でSMARCA2の病的バリアント(表1参照)のヘテロ接合が同定されることにより確定する。
用いられる分子遺伝学的検査としては、単一遺伝子検査、マルチ遺伝子パネルの使用、網羅的ゲノム検査などが考えられる。
- 単一遺伝子検査
最初にSMARCA2の配列解析を行い、そこで病的バリアントが検出されなかった場合は、遺伝子標的型欠失/重複解析に進む。
- マルチ遺伝子パネル
SMARCA2をはじめとする関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネルの使用が考えられる。
注:
(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、今このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
したがって、どのマルチ遺伝子パネルを用いれば、意義不明のバリアントや現状の表現型と無関係な病的バリアントの検出を抑えつつ、いま問題にしている疾患の遺伝的原因を特定できる可能性が高いかという点について、臨床医の側であらかじめ検討しておく必要がある。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝子検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
- 網羅的ゲノム検査
Nicolaides-Baraitser症候群の症候を呈する症例であるにもかかわらず、単一遺伝子検査(ないし、SMARCA2を含むマルチ遺伝子パネルの使用)で診断が確定しないような場合は、網羅的ゲノム検査も視野に入る。
具体的には、エクソームシーケンシングとゲノムシーケンシングが考えられる。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝子検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
表1:Nicolaides-Baraitser症候群で用いられる分子遺伝学的検査
| 遺伝子1 | 手法 | その手法で病的バリアント2が検出される発端者の割合 |
|---|---|---|
| SMARCA2 | 配列解析3 | 36/444 |
| 遺伝子標的型欠失/重複解析5 | 2/616 | |
| 不明7 | 対象外 | 不明 |
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- この遺伝子で検出されているアレルのバリアント内容に関する情報については、「分子遺伝学」の項を参照のこと。
- 配列解析を行うことで、benign、likely benign、意義不明、likely pathogenic、pathogenicといったバリアントが検出される。
- Van Houdtら[2012]
- 遺伝子標的型欠失/重複解析では、遺伝子内の欠失や重複が検出される。
- Tsurusakiら[2012],Wolffら[2012]
- NCBRSを示唆する症候を呈するにも関わらず、SMARCA2の病的バリアントが検出されない例については、SWI/SNFクロマチンリモデリング複合体に属する他の遺伝子が原因として関与する座位異質性の存在が、可能性として考えられる。
バリアントの種類としては、遺伝子内の小さな欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。
配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失/重複の検出を目的に設計された遺伝子標的型マイクロアレイなど、さまざまなものがある。
Van Houdtら[2012]が評価を行った44人のうち、NCBRSの臨床診断が確実視されていたのは37人のみで、そのうち34人にSMARCA2の病的バリアントが検出されている。
残りの3人についてはARID1Bの病的バリアントが検出され、Coffin-Siris症候群への再分類が行われている(「鑑別診断」の項を参照)。
臨床的特徴
臨床症状
Nicolaides-Baraitser症候群(NCBRS)でSMARCA2の病的バリアントが確認されている61例(男性35人,女性26人)において、最も多くみられた症候を表2にまとめた[Sousaら2014]。
表2: Nicolaides-Baraitser症候群罹患者61人に多くみられた臨床症候の概要
| 症候 | それが現れる罹患者の割合 |
|---|---|
| 知的障害 | 100% |
| 疎な毛髪 | 97% |
| 指節間関節の膨隆 | 85% |
| 粗野な顔 | 77% |
| 小頭症 | 65% |
| 癲癇発作 | 64% |
Sousaら[2014]より
外胚葉
頭髪は出生時より疎であることが多いものの、年齢とともに、特に10歳代にこれがより顕著になっていく。
ただ、疎さの程度が経時的に改善していった例もみられる。
皮膚は、色素沈着の程度が低いように見受けられるものの、真性の皮膚白皮症を有しているわけではない。
皮下脂肪が少ないため、静脈が目立ち、指節間関節が膨隆する。
末節骨の幅は年齢とともに広くなり、幅広の楕円形を呈するようになる。
経時的に第1趾・第2趾間の空隙拡大がみられるようになることがある。
歯の萌出遅延が多くみられ、永久歯の萌出誘導を目的とした先行乳歯の抜歯が必要になることがしばしばある。
癲癇発作
癲癇発作は管理が難しく、ある程度のコントロールを達成するには複数の抗痙攣薬が必要になることがある。
発作の初発年齢は、平均で24ヵ月、中央値で18ヵ月である。
分布範囲は出生時から14歳までである。
発作の種類は、さまざまなものが報告されている。
発作の初発に伴い、発達の進行に、退行や停止がみられたとする報告がみられる。
発達遅滞/知的障害
罹患者の半数近くが重度の発達遅滞/知的障害(ID)を示し、特にスピーチや言語の発達に顕著な遅れがみられる。
3分の1は中等度の知的障害を、残りが軽度の知的障害を示す。
3分の1近くの罹患者は、スピーチ・言語技能の獲得に至らない。
自立歩行の開始年齢は、平均21ヵ月(範囲は10ヵ月-5歳)である。
精神運動機能の退行はそれほど多くはないものの、発作が多発し、それが次第に悪化していく例については、スピーチの機能が喪失することが多いとされている。
行動上の問題は、少なくとも19人で報告されており、その一部に、自閉症様症候(固執,聴覚過敏等)を示す例が確認されている。
ただ、文献にある範囲では、NCBRS罹患者で、正式に自閉症との診断が下りた例はみられない。
成長
罹患者の約半数(24/46)は低出生体重で、低身長も同じ割合でみられる。
身長については、50パーセンタイルを超えた例はみられない。
小頭症は後天性の傾向がみられ、出生時には3分の1近く(7/30)、その後のフォローの段階では3分の2(34/52)で認められている。
他の所見
- 男性罹患者の大多数に停留精巣がみられる。
- 難聴は、罹患者59人中4人で報告されている。
- 視覚障害は、近視が10例、乱視が4例に認められている。
- 先天性心疾患(例えば、心房中隔欠損,肺動脈狭窄,大動脈縮窄,動脈管開存,重複大動脈弓)が6人の罹患者に報告されている。
難聴の本態や発症年齢に関する報告はみられない。
遺伝型-表現型相関
明確な遺伝型-表現型相関はみられないものの、ATPアーゼドメインのC末端ヘリカーゼ領域内に病的バリアントを有する全例が重度の知的障害、癲癇を示し、この遺伝子の他の部位に病的バリアントを有する例より割合が高かった。
浸透率
浸透率を確認するに足る十分なデータが揃っていない現状である。
現在までに報告されている61例すべてが新生の病的バリアントに起因するものであることから、浸透率は100%であるように思われる[Sousaら2014]。
命名法について
わかっている範囲で最も早くNCBRSを報告した1993年の論文の著者2人の名に因んで、Morinら[2003]がNicolaides-Baraitser症候群という名称を提唱した。
発生頻度
NCBRSの発生頻度はよくわかっていないが、きわめて低いものと推定される。
文献の形で報告された罹患者の数は100人に満たない。
遺伝子の上で関連のある疾患(同一アレル疾患)
SMARCA2の病的バリアントに関連して生じるものとしては、このGeneReviewで述べたもの以外の表現型は知られていない。
本症候群の症候を一切有さず、腫瘍単発の形で現れる孤発性腫瘍において、しばしばSMARCA2の体細胞バリアントをもつ例がみられる。
ただ、このバリアントは生殖細胞系列にみられるわけではないので、こうした腫瘍に関する素因に継承性がみられるということはない。
鑑別診断
Coffin-Siris症候群(CSS)は、粗野な顔、多毛症、第5指趾の爪の欠損ないし低形成、脳梁無形成、発達遅滞/知的障害を特徴とする疾患である。
顔貌についてはNicolaides-Baraitser症候群と類似することもあるが、NCBRS罹患者の場合は指節間関節の膨隆という特徴的症候を示す一方、第5指趾の低形成は有しない。
そのため、両疾患を鑑別する上では、指趾の所見が非常に重要な意味をもつ。
加えて言うと、NCBRS罹患者の頭髪が疎であるのに対し、CSSでは多毛症を示す。
CSSは、ARID1A、ARID1B(「ARID1B関連疾患」のGeneReviewを参照)、SMARCA4、SMARCB1、SMARCE1のいずれか1つのヘテロ接合性病的バリアントに起因して生じる。
罹患者の大多数については、そうしたバリアントが新生の形で生じたものである。
CSSとNCBRSの間に表現型の重なりがみられる理由は、おそらく、両疾患ともSWI/SNF複合体関連遺伝子の病的バリアントに起因するという共通点にあるように思われる。
注目すべき報告として、Tsurusakiら[2012]が、Coffin-Siris症候群(CSS)の臨床診断を受けた1例について、SMARCA2の55kbの中間部欠失が判明し、その臨床症候がCSSよりもNCBRSに一致したもの(第5指趾の爪の欠損はみられず、指節間関節の膨隆がみられた)であったとの報告がある。
Williams症候群(WS)は、典型的顔貌(長い人中,厚い上下赤唇,膨らんだ頰)、大動脈弁上狭窄、関節弛緩、成長障害、高カルシウム血症、知的障害を特徴とする。
ただ、認知プロファイルを見ると、言語面に相対的な強みがみられる。
WSとNCBRSとの鑑別は、顔貌、心疾患、認知プロファイルだけで十分である。
WSは、Williams-Beuren症候群関連領域(WBSCR)の微小欠失に起因して生じる。
罹患者の大多数は、病的バリアントが新生の形で生じたものである。
Cornelia de Lange症候群は、典型的顔貌(長い人中,アーチ状の眉、眉毛癒合,短い鼻)、成長障害、四肢奇形を特徴とする。
四肢の症候は基本的に指趾の低形成を伴うもので、時に重篤な例もみられる。
CdLSとNCBRSの鑑別には、顔面症候と四肢の変化だけで十分である。
CdLSは、NIPBL、HDAC8、RAD21、SMC1A、SMC3のいずれか1つの遺伝子のバリアントに起因して生じる。
罹患者の大多数は新生の病的バリアントに起因するものである。
2q37微小欠失症候群は、典型的顔貌(丸い顔,前額部の突出,アーチ状の眉、眼瞼裂斜上,内眼角贅皮,低発達の鼻翼,薄い上赤唇)、第3-4指趾の中手(足)骨-指節骨短縮、低身長、筋緊張低下、知的障害を特徴とする。
2q37欠失症候群罹患者で癲癇発作の報告がみられるものの、通常、管理はそれほど難しくはない。
大多数の例は、新生の形で生じた微小欠失である。
ビオチニダーゼ欠損症
ビオチニダーゼ欠損症では、基本的に、癲癇発作、筋緊張低下、毛髪の喪失を含む皮膚の異常が現れる。
ふつう、顔貌は正常である。
ビオチニダーゼ欠損症は、BTDの両アレルの病的バリアントに起因して生じ、常染色体潜性の遺伝形式をとる。
臨床的マネジメント
最初の診断に続いて行う評価
Nicolaides-Baraitser症候群(NCBRS)と診断された罹患者については、疾患の範囲やニーズを把握するため、以下のような評価を行うことが推奨される。
- 癲癇発作の有無を確認するための脳波検査。
- 発達指標に関する評価、ならびに神経学的問題の有無や状況を確認するための神経学的診査や発達診査。
- 必要に応じ、作業療法、言語治療、理学療法方面の評価。
- 歯の萌出に遅延がみられるようであれば、歯科的評価。
- 難聴の評価を目的とした聴覚脳幹反応検査や耳音響反射検査を用いた聴覚評価。
- 視力検査や散瞳眼底検査を含む眼科的診査。
- 男性については停留精巣の評価。
- 臨床遺伝医ないし遺伝カウンセラーとの面談。
その上で、発作の管理を目的として癲癇専門医への紹介を検討する。
症状に対する治療
以下のような形にするのが適切である。
- 癲癇発作に対しては、神経内科医もしくは癲癇専門医の管理下で抗痙攣薬(ASM)の投与。
- 発達を理想的な形で誘導するための作業療法、理学療法、言語治療。
- 必要に応じ、屈折異常を矯正するための眼鏡の使用。
- 必要に応じ、補聴器の使用。
- 必要に応じ、精巣固定術。
定期的追跡評価
以下の内容で定期的追跡評価を行う。
- 癲癇発作の評価や管理を目的とした、神経内科医による少なくとも年に1度の評価。
- 発達の進行状況、治療的・教育的介入の評価を目的とした、発達小児科医による年に1度の評価。
- 眼科的異常、聴覚的異常に関する定期的フォロー。
リスクを有する血縁者の評価
遺伝カウンセリングを目的として、リスクを有する血族に対して行う検査関連のことについては、「遺伝カウンセリング」の項を参照のこと。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「Clinical Trials.gov」、ならびにヨーロッパの「EU Clinical Trials Register」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
Nicolaides-Baraitser症候群(NCBRS)は、常染色体顕性の遺伝形式を示す。
現在までに報告されている罹患者は、すべて新生の病的バリアントに起因して生じたものである。
家族構成員のリスク
発端者の両親
- 現在までに報告されているNCBRSの発端者は、すべて、SMARCA2に生じた新生の病的バリアントに起因する例である。
- 見かけ上、新生の病的バリアントと思われる発端者については、その両親に対して、発端者で検出されたSMARCA2の病的バリアントに関する検査を行うことが推奨される。
しかしながら、本疾患については高い浸透率が見込まれることから、見かけ上、無症候と思われる人から病的バリアントが発見される可能性はきわめて低いものと推察される。
発端者の同胞
現在のところ、罹患者はすべてSMARCA2の新生の病的バリアントに起因する例であることから、同胞に対するリスクは低いものと推察される。
ただ、理論上は片親の生殖細胞系列モザイクの可能性が残っていることから、同胞に関する再発危険率はおおむね1%と考えられる。
発端者の子
NCBRS罹患者の子はそれぞれ、SMARCA2の病的バリアントを継承する50%の可能性を有する。
ただ、現在のところ、NCBRS罹患者が子を儲けたという報告はみられない。
他の家族構成員
現在までに報告されているNCBRSの発端者は、全員、SMARCA2の新生の病的バリアントに起因する例であるため、他の血族の有するリスクは低いように思われる。
遺伝カウンセリングに関連した問題
家族計画
- 遺伝的リスクの確定や、出生前/着床前遺伝子検査を受けるかどうかの話し合いといったことに最も適しているのは、妊娠前の時期である。
- 罹患者の両親に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法であるとか、遺伝子・病原のメカニズム・疾患等に対するわれわれの理解は、将来、より進歩していくことが予想される。
そのため、分子的な診断が確定していない(すなわち、原因となった遺伝的メカニズムがわかっていない)罹患者のDNAについては、保存しておくことを検討すべきである。
出生前検査ならびに着床前の遺伝子検査
家系内に存在するSMARCA2の病的バリアントの内容が同定されている場合は、高リスクの妊娠に備えた出生前診断や、NCBRSを対象とした着床前遺伝子検査を行うことが可能となる。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
-
Genetic and Rare Diseases Information Center (GARD)
Nicolaides-Baraitser syndrome
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:Nicolaides-Baraitser症候群:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specific データベース |
HGMD | ClinVar |
|---|---|---|---|---|---|
| SMARCA2 | 9p24.3 | Probable global transcription activator SNF2L2 | SMARCA2 @ LOVD | SMARCA2 | SMARCA2 |
データは、以下の標準資料から作成したものである。
遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。
リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:Nicolaides-Baraitser症候群関連のOMIMエントリー(閲覧はすべてOMIMへ)
| 6000014 | SWI/SNF-RELATED, MATRIX-ASSOCIATED, ACTIN DEPENDENT REGULATOR OF CHROMATIN, SUBFAMILY A, MEMBER 2;SMARCA2 |
| 601358 | NICOLAIDES-BARAITSER SYNDROME;NCBRS |
分子レベルの病原
ATPアーゼ依存性クロマチンリモデリング因子に属するSWI/SNFファミリーは、遺伝子発現、分化、発生といったものの制御に必要不可欠な役割を果たしている。
SMARCA2は、SMARCA2をコードしている。
SMARCA2は、ATP加水分解とDNA結合とをカップリングさせてクロマチンリモデリングを生じさせる上で必要な、ATPアーゼドメインを共有するヘリカーゼ関連タンパク質ファミリーの一員である。
SMARCA2は、SWI/SNF複合体のヒトアナログであるBRG1関連因子(BAF)複合体の1つのサブユニットである。
Nicolaides-Baraitser症候群(NCBRS)を示唆する症候を有しつつも、SMARCA2に病的バリアントが検出されない例については、これまで、SWI/SNF複合体内の他の遺伝子のバリアントが原因として関与していることが示唆されている[Van Houdtら2012]。
遺伝子構造
SMARCA2は、34のエクソンを有し、5,879塩基対から成る転写産物を産生する。
この遺伝子は、選択的スプライシングにより、CAGトリヌクレオチドリピートの長さに多型を有する、複数のアイソフォームをコードする転写産物のバリアントを産生することがわかっている。
遺伝子ならびにタンパク質の情報に関する詳細は、表Aの「遺伝子」の欄を参照されたい。
病的バリアント
NCBRS罹患者61人のレビューで、59人が病的ミスセンスバリアントを、2人がインフレームの欠失を有していた[Sousaら2014]。
病的バリアントはすべて、SMARCA2のATPアーゼドメイン(エクソン15-25)に集中していた。
こうした知見に基づいて、SMARCA2の病的バリアントは、ドミナントネガティブないし機能獲得型の作用を引き起こしている可能性が高いものと考えられている。
正常遺伝子産物
SMARCA2は、ATPアーゼ依存性クロマチンリモデリング因子に属するSWI/SNFファミリーの中の、1,590個のアミノ酸から成る触媒サブユニットをコードしている。
この触媒サブユニットには、ヘリカーゼ様ATPアーゼドメインがあって、ATP結合やATP加水分解とDNA結合とをカップリングすることで遺伝子発現を制御する役割を担っている。
異常遺伝子産物
SMARCA2に病的バリアントが生じることで、結果的にクロマチンリモデリングの異常が生じ、下流にある別の遺伝子の制御異常を引き起こしてNCBRSの表現型に至るものと考えられている。
変異によって産生されたSMARCA2は、SWI/SNF複合体中に組み込まれる能力を保持し、ゲノム内にある下流の標的に結合することになるのではないかと考えられている。
このシナリオに従えば、ドミナントネガティブ効果ないし機能獲得型効果により、遺伝子発現に必要なヌクレオソーム構造のリモデリングが正常な形で生じなくなっているということになる。
更新履歴:
-
Gene Reviews著者: Omar Abdul-Rahman, MD
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2015.10.15. 日本語訳最終更新日: 2022.12.3.[in present]
![]()