鏡‐緒方症候群
(Kagami-Ogata Syndrome)
[Synonyms: KOS14]
Gene Review著者: Tsutomu Ogata, MD, PhD and Masayo Kagami, MD, PhD.
日本語訳者: 緒方勤(浜松医科大学医学部)
Gene Review 最終更新日: 2024.10.24. 日本語訳最終更新日: 2025.12.10
要約
疾患の特徴
鏡-緒方症候群は、発達遅滞、知的障害、嚥下障害を伴う摂食困難、ふっくらとした頬、突出し深い人中、コートハンガー様肋骨を伴うベル型小胸郭、および腹壁欠損(臍帯ヘルニアや腹直筋離開)を特徴とする。その他の一般的な特徴には、関節拘縮、後側弯症、外反股、および喉頭軟化症が含まれる。また、心疾患や肝芽腫の報告もある。
診断・検査
鏡-緒方症候群の診断は、示唆的な症状と以下のいずれかによって母親由来RTL1アンチセンス(RTL1as)の発現低下を示唆する遺伝学的検査結果が得られた場合に確定する
- 父親由来の第14染色体の単親性ダイソミー(upd(14)pat)
- 通常はメチル化されていない母親由来MEG3/DLK1:IG-DMRおよびMEG3:TSS-DMRのエピ変異(過剰メチル化)
- MEG3/DLK1:IG-DMRおよび/またはMEG3:TSS-DMRやRTL1asを含む母親由来の14q32.2領域の欠失
- MEG3/DLK1:IG-DMRやMEG3:TSS-DMRを含まない母親由来RTL1asの欠失
- 母親由来MEG3:TSS-DMRにあるMEG3プロモーターとRTL1asの連続性を破断する転座(または逆位)
臨床的マネジメント
治療:
- 発達および教育の支援
- 必要に応じた出生後の人工呼吸管理および酸素療法
- 必要に応じた気管切開
- 上・下気道感染の監視と治療
- 側弯症の整形外科的治療
- 関節拘縮のリハビリテーション治療
- 低体重改善のための経管栄養(必要に応じて胃ろう)
- 摂食訓練
- 心疾患の治療(循環器科による対応)
- 肝芽腫の標準治療(外科手術および化学療法)
- 社会福祉支援および家族支援
サーベイランス:
- 発達の進行、教育的ニーズ、誤嚥や呼吸不全の兆候、後弯弯症および関節拘縮の進行、栄養状態、経口摂取の安全性、便秘の有無、家族やケアコーディネーションの必要性を各診察時に確認
- 心エコー検査を毎年実施
- 腹部超音波検査および血清α-フェトプロテイン(AFP)検査を3〜4歳まで3か月ごとに実施
避けるべき事項:
- 呼吸器感染症の曝露を避ける
- 鏡-緒方症候群の患者は特に乳児期において、呼吸器感染症による呼吸不全を発症するリスクが高い
遺伝カウンセリング
鏡-緒方症候群の再発リスクは、母親由来のRTL1asアレルの発現低下を引き起こす遺伝的メカニズムに依存する。ほとんどの患者では、遺伝的要因が新生突然変異として発生するため、同胞への再発リスクは増加しない。ただし、まれに、原因となる遺伝的異常を持つ親が存在し、同胞への再発リスクが高くなる場合がある。母親由来の欠失を伴う場合、同胞における発症が報告されている。発端者が14q32.2領域の欠失や転座を有する、出生前診断や着床前遺伝子検査が可能である。胎児DNAのメチル化検査(MEG3/DLK1:IG-DMRおよびMEG3:TSS-DMRの異常メチル化の検出)は推奨されない。羊水由来のDNAが最も信頼性の高い組織検体と考えられているが、偽陰性の報告があるためである。
診断
鏡-緒方症候群に関する統一された臨床診断基準は確立されていない。
示唆的所見(診断の手がかり)
鏡-緒方症候群は、以下の臨床所見を示す患者において疑われるべきである。特に、特異的(診断的価値が高い)な所見に加えて、特徴的だが特異的ではない所見および非特異的所見を伴う場合に診断の可能性が高まる【Kagami et al 2015, Ogata & Kagami 2016】。
特異的(診断的価値が高い)所見
- ふっくらとした頬、および突出した深い人中(図1参照)。
- コートハンガー様肋骨を伴うベル型小胸郭(図2参照)。
注:コートハンガー肋骨角は、胎児期中期から小児期にかけて増大する。胸郭の中間径と最大径の比率は、出生時から幼児期にかけて低下する【Miyazaki et al 2011, Kagami et al 2015, Ogata & Kagami 2016, Kuriki et al 2022】。

図 1.
母親由来のDLK1、MEG3/DLK1:IG-DMR、MEG3:TSS-DMR、MEG3、RTL1/RTL1as、MEG8、およびセントロメア側の一部snoRNAを含む微小欠失によるKagami-Ogata症候群の女児。乳児期、生後2年、8歳時の様子。乳児期から小児期を通じて、特徴的なふっくらとした頬および目立つ深い人中が観察される。Ogata & Kagami [2016] より改変。
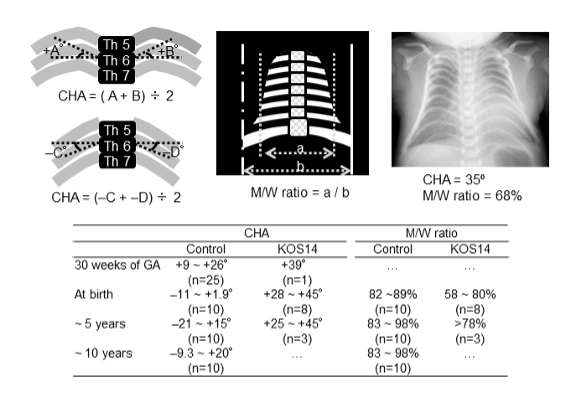
図2.
Kagami-Ogata症候群の日本人新生児の胸部X線写真。コートハンガー角(CHA)は妊娠30週から5歳までの間に増加する。CHAは、第6肋椎関節部を基準とした水平軸と、第6後方肋骨の頂点との間の右および左の角度の平均である。注:肋骨が下向きに傾いている場合は、第6肋骨の中点をCHAの測定に使用する。胸郭の中間径と最広部径の比率(M/W比)は、Kagami-Ogata症候群の乳児および幼児で低下する。Kagamiら [2015] および Ogata & Kagami [2016] より改変。
特徴的だが特異的ではない所見
- 腹壁欠損(臍帯ヘルニア、腹直筋離開 など)
- 胎盤過形成
- 羊水過多
非特異的所見
- 頭蓋顔面および頸部の特徴(前頭突出、前額多毛、眼裂狭小、低い鼻梁、前向きの鼻孔、しわの寄った尖った唇、小顎、短く翼状様の頸部)
- 出生体重が相対的に大きい
- 中等度から重度の発達遅滞
- 嚥下障害を伴う摂食困難
- 関節拘縮
- 後側弯症
注:特徴的顔貌、コートハンガー様肋骨を伴うベル型小胸郭、および臍帯ヘルニアは、胎児超音波検査およびMRIにより妊娠第2三半期から確認可能である【Chen et al 2019, Igreja da Silva et al 2019, Molinet Coll et al 2021, Kuriki et al 2022】。
診断の確立
鏡-緒方症候群の診断は、示唆的所見を持つ患者において、以下のいずれかの遺伝的異常により母親由来RTL1asの発現低下が確認された場合に確定する(表1参照)。
- 第14番染色体父性片親性ダイソミー(upd(14)pat):2つの第14番染色体が共に父に由来する状態(約50%の患者に認められる)。
- エピ変異:通常低メチル化を示す母親由来MEG3/DLK1 intergenic differentially methylated region(MEG3/DLK1:IG-DMR)およびMEG3 transcription start site DMR (MEG3:TSS-DMR)の高メチル化(約25%の患者に認められる)。
- 母親由来14q32.2インプリンティング領域の欠失:MEG3/DLK1:IG-DMRおよび/またはMEG3:TSS-DMRを含み、RTL1asを含むこともある(約20%の患者に認められる)。
- 母親由来RTL1asの欠失:RTL1asを含み、MEG3/DLK1:IG-DMRやMEG3:TSS-DMRは含まない欠失(約5%の患者に認められる)。
- 転座または逆位: 母親由来のMEG3:TSS-DMRにあるMEG3プロモーターとRTL1asの連続性が破断されている状態(まれ)。
遺伝学的検査
鏡-緒方症候群の遺伝学的検査には、推奨される第一段階の検査(MS-MLPA)と推奨される第二段階の検査(第14番染色体の由来親検査)があり、これにより遺伝学的原因と反復リスクが評価される。
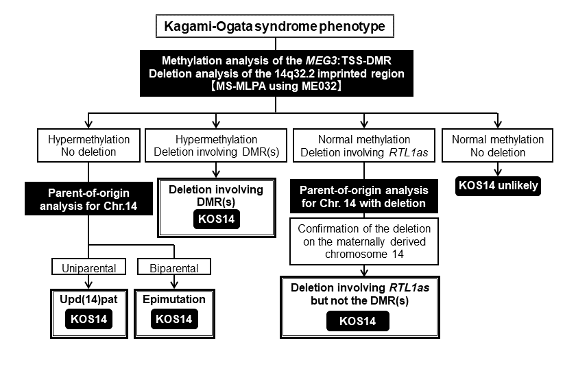
図 3.
Kagami-Ogata症候群の診断および分子的原因を明らかにするための検査アルゴリズム。
第一段階の検査
MS-MLPAは、MEG3:TSS-DMRのメチル化解析とMEG3/DLK1:IG-DMR、MEG3:TSS-DMR、RTL1asを含む欠失解析を同時に行える方法である。この検査は、upd(14)patやエピ変異による異常メチル化と、欠失による異常メチル化を区別できるため、再発リスクの評価に重要である。なお、この検査はMEG3/DLK1:IG-DMRの欠失を検出できるが、MEG3/DLK1:IG-DMRのメチル化異常は検出できない。
- メチル化異常(MEG3:TSS-DMRの過剰メチル化)があり、欠失がなければ、第二段階の検査(第14番染色体の由来親検査)により、upd(14)patとエピ変異が鑑別できる。
- メチル化異常がなく、RTL1asを含む欠失が検出されれば、第二段階の検査により、RTL1asを含む欠失由来親を判定できる。
第二段階の検査
第14番染色体の由来親解析:DNA多型を患者および両親のDNAサンプルを用いて検査するもので、、upd(14)patと両親性第14番染色体を伴うエピ変異を示唆を区別できる。なお、upd(14)patが確認された場合、ロバートソン転座やi(14q)染色体を同定し、再発リスクを評価するために核型検査が推奨される【Ogata & Kagami 2016】。
RTL1as欠失の親由来解析:母親由来のRTL1as欠失または母親由来第14染色体に生じた新生欠失が鏡-緒方症候群の原因となる。
その他の検査(推奨されない)
- メチル化解析(メチル化特異的PCR、バイアサルフィトシーケンス、パイロシーケンス):14q32.2領域の異常メチル化パターンを検出できるが、欠失とupd(14)pat・エピ変異を区別できない。
- 高密度SNPアレイ解析:小さな欠失や14q32.2の父性片親性親イソダイソミーは検出可能だが、父性片親性親ヘテロダイソミーは検出できない。
表1. 鏡-緒方症候群における分子遺伝学的検査
| 方法 | 遺伝的メカニズム1 | この方法で検出される患者の割合2 |
|---|---|---|
| MS-MLPA3 | 第14番染色体父性片親性ダイソミー | 約50% |
| 母親由来MEG3:TSS-DMRのエピ変異(過剰メチル化) | 約25% | |
| 母親由来MEG3/DLK1:IG-DMRおよび/またはMEG3:TSS-DMRの欠失 | 約20% | |
| 母親由来RTL1asの欠失4 | 約5% 5 | |
| 核型分析(カリオタイピング) | MEG3プロモーターとRTL1asの連続性を破断する転座や逆位 | 極めて稀6 |
| 第14染色体を含むロバートソン転座 | 稀 |
略語:
DMR = differentially methylated region
IG = intergenic
MS-MLPA = methylation-specific multiple ligation-dependent probe amplification
TSS = transcription start site
upd(14)pat = paternal uniparental disomy of chromosome 14
注釈
- 詳細は分子遺伝学の項および図4を参照。
- 出典: Ogata & Kagami [2016]、Omark et al. [2021]、Mackay et al. [2022]、Eggermann et al. [2023]。
- MEG3:TSS-DMRの過剰メチル化が確認されれば、鏡-緒方症候群の診断が確定する。 ただし、欠失解析(MS-MLPAを含む)および由来親解析が、upd(14)pat・エピ変異・DMR欠失を区別するために必要である。
- RTL1as欠失が母親由来14番染色体に存在することを確認するためには親由来解析が必要である。
- MEG3/DLK1:IG-DMRやMEG3:TSS-DMRの欠失を伴わないRTL1asの欠失は、鏡-緒方症候群の5人の患者で確認されている。ただし、このような欠失は、稀な状態にたいする報告バイアスにより過大評価されている可能性がある。
- 母由来MEG3:TSS-DMR内のMEG3プロモーターとRTL1asの間の構造が破断される転座は、鏡-緒方症候群の1例で確認されている。
臨床像
臨床的記述
鏡-緒方症候群は、以下の特徴を示す疾患である:
- 発達遅滞
- 知的障害
- 摂食困難
- ふっくらとした頬、目立つ深い人中
- 小さく鐘型の胸郭(コートハンガー状の肋骨を伴う)
- 腹壁欠損(臍帯ヘルニア、腹直筋離開)
- 関節拘縮
- 後弯側弯症(キホスコリオーシス)
- 寛骨臼形成不全(コクサ・ヴァルガ)
- 喉頭軟化症
- 心疾患
- 肝芽腫
現在までに約100人の患者が鏡-緒方症候群と診断されている【Sakaria et al 2021, Mackay et al 2022, Smith et al 2024, Ogata T & Kagami M, Unpublished observation】。
表2. 鏡-緒方症候群の主な特徴と頻度
| 特徴 | 該当患者の割合 | コメント |
|---|---|---|
| 妊娠・分娩 | ||
| 羊水過多 | >95% | |
| 胎盤肥大 | 約85% | 胎盤サイズが正常の120%以上 |
| 発達・認知 | ||
| 発達遅滞 | >95% | 中等度~重度 |
| 知的障害 | 100% | |
| 栄養・成長 | ||
| 摂食困難 | >95% | |
| 出生前の過成長 | >50% | 出生時身長および/または体重が平均の+2SD以上 |
| 出生後の成長障害 | 約35% | 身長および/または体重が平均の-2SD以下 |
| 頭蓋顔面の特徴 | ||
| ふっくらとした頬、目立つ深い人中 | >90%-95% | 最も一般的で特異的な特徴 |
| その他 | 前頭突出、多毛性前額、睫裂狭小、低い鼻梁、前転した鼻孔、狭く突き出た唇、小顎、短く幅広い頸部 | |
| 骨格異常 | ||
| 小さく鐘型の胸郭 | 100% | |
| コートハンガー状肋骨 | 100% | |
| 関節拘縮 | 60%-65% | |
| 後弯側弯症(キホスコオーシス) | 約40% | |
| 寛骨臼形成不全(コクサ・ヴァルガ) | 約33% | |
| 呼吸器系 | ||
| 喉頭軟化症 | 約40% | |
| 腹壁欠損 | ||
| 腹直筋離開 | 65%-70% | |
| 臍帯ヘルニア | 約30% | |
| その他の特徴 | ||
| 心疾患 | 25% | |
| 肝芽腫 | 5%-10% |
略語:
SD = 標準偏差(standard deviation)
出典:
Kagami et al [2015]; 類似データはSakaria et al [2021]にも要約されている。
妊娠と分娩
羊水過多は通常、妊娠第2三半期、中央値25.5週(範囲14~30週)で同定される。
妊娠25週以降では約80%の妊婦で羊水除去が必要となり、30週以降ではほぼ全例で実施される。
羊水過多の原因は胎盤肥大および嚥下障害によるものである。
胸郭および腹部の異常は、妊娠約25週の時点で胎児超音波検査により約40%で検出される。
早産は**頻繁に発生し(約80%)、出産時の中央値は32.5週(範囲30~35週)**である。
発達遅滞
- 定頸:中央値7か月(範囲3~36か月)
- 支えなしの座位獲得:中央値12か月(範囲8~25か月)
- 支えなしの歩行獲得:中央値25.5か月(範囲20~49か月)
軽度の遅れのみが認められた症例は2例あり、**1例はエピ変異を有する患者【Higashiyama et al 2022】、もう1例はモザイク型upd(14)patの患者【Haug et al 2017】**であった。
知的障害
- 知的障害はすべての患者に認められる。
- **IQの中央値は55(範囲29~70)**である。
脳MRIの異常所見は、検査を受けた5人の患者では確認されていない。
成長
- 出生前の成長は良好であり、出生体重および身長は通常平均以上である。
- 出生体重の範囲:-0.1SDから+8.8SD
- 出生身長の範囲:-1.7SDから+3.0SD
しかし、出生後の成長はしばしば低下し、主に呼吸不全および摂食困難による栄養不良が原因とされる。
- 1~15歳の時点での体重範囲:-6.0SDから+4.0SD
- 1~15歳の時点での身長範囲:-8.7SDから+1.1SD
頭蓋顔面の特徴(図1参照)
鏡-緒方症候群の患者では以下の特徴が認められる:
- 前頭突出(約75%)
- 多毛性前額(約70%)
- 睫裂狭小(70~75%)
- ふっくらとした頬(>90%)
- 低い鼻梁(>90%)
- 前転した鼻孔(80%)
- 目立つ深い人中(>95%)
- しわの寄った(やや狭くテント状の)唇(>50%)
- 小顎(~100%)
- 短く幅広い頸部(>90%)
これらの顔貌の特徴は乳児期に既に認められ、小児期を通じて一貫して確認できる。
多くの特徴は非特異的であるが、「ふっくらとした頬」と「目立つ深い人中」は鏡-緒方症候群に特異的な特徴と考えられる。
胸郭異常と呼吸不全
小さく鐘型の胸郭は乳児期に明確に認められる。
コートハンガー状の肋骨の角度増加、および胸郭中間径と最大径の比率低下は、胎児期から小児期にかけて客観的に評価可能(図2参照)。
胸郭の形態は乳児期以降では正常となるが、コートハンガー状肋骨は小児期を通じて認められる。
胸郭の異常により、90%以上の患者で人工呼吸管理が必要となり、その中央値は1か月(範囲0.1~17か月)である。
重度の喉頭軟化症を伴う場合、気管切開が必要となることがある。
その他の骨格異常
- 関節拘縮
- 後弯側弯症(キホスコリオーシス)
- 寛骨臼形成不全(コクサ・ヴァルガ)
患者ごとに重症度のばらつきがある。
摂食・消化器系の症状
- 生後1週間以上生存したすべての患者で経管栄養が必要であり、その中央値は7.5か月(範囲0.1~89か月)である。
- 腹直筋離開は一般的であり、臍帯ヘルニアも発生しうる。
- 腹壁の筋力低下により便秘を引き起こすことがある。
心疾患
- 約25%の患者に心血管奇形が認められる。
- 確認された8例のうち:
- 心房中隔欠損(2例)
- 心室中隔欠損(1例)
- 動脈管開存症(4例)
- 肺動脈狭窄(1例)
これらの心疾患は軽度であり、強力な薬物治療や手術は不要とされる。
心筋症や伝導障害の報告はない。
肝芽腫
これまでに報告された100例のうち、3例で乳児期に肝芽腫が確認されている。
その他
- 単純部分発作(simple seizure)が1例報告されている【Kagami et al 2015】。
予後
- 5歳未満の死亡率は比較的高く、20~25%の患者が死亡する。
- しかし、5歳以上の患者においては死亡率の上昇は認められない。
- 死因は様々であるが、主な原因は呼吸不全である。
現在までに18歳以上の成人患者が9名確認されており、最年長の患者は35歳である【Smith et al 2024】。
遺伝型-表現型の相関
- 明確な遺伝型-表現型相関は特定されていない【Kagami et al 2015】。
- upd(14)pat、エピ変異、およびRTL1asを含まない欠失では、対照群の約5倍のRTL1発現が認められる。
- RTL1asを含む欠失では、対照群の約2.5倍のRTL1発現が確認されている。ただし、RTL1asを含む欠失の報告例が少ないため、これが表現型の軽症化と関連するかは不明である。
浸透率(Penetrance)
浸透率は完全であり、すべての罹患者に以下の特徴が認められる。
- ふっくらとした頬
- 目立つ深い人中
- 小さく鐘型の胸郭(コートハンガー状の肋骨を伴う)
ただし、分子遺伝学的検査は臨床的特徴を持つ個体に対してのみ実施される傾向があるため、診断の偏りが存在する可能性がある。実際、これまでにupd(14)patのモザイクを持つ2人の患者が報告されており、1例は非常に軽度な鏡-緒方症候群の表現型を示し、もう1例は典型的な症候群の特徴を有していた【Haug et al 2017, Li et al 2021】。そのため、正常に近い表現型を持つモザイク患者は見逃されている可能性がある。
命名法(Nomenclature)
鏡-緒方症候群は、かつてupd(14)pat症候群と呼ばれていたことがある。しかし、鏡-緒方症候群は必ずしも父親由来の第14染色体単親性ダイソミー(upd(14)pat)のみで発症するわけではなく、他の遺伝的メカニズムによっても引き起こされるため、「upd(14)pat症候群」という名称は誤解を招く可能性がある。そのため、第14染色体q32.2インプリンティング領域の(エピ)遺伝的異常によって引き起こされるこの独特な臨床疾患を表すために、「鏡-緒方症候群(Kagami-Ogata syndrome)」という名称が提唱された。
有病率(Prevalence)
これまでに約100例の鏡-緒方症候群の患者が報告されている【Mackay et al 2022, Eggermann et al 2023】。日本では、報告された症例を含めて76例の患者が確認されている【Kagami & Ogata, personal observation】。診断されていない患者が多数存在する可能性が高い。
遺伝的に関連する疾患(Genetically Related (Allelic) Disorders)
テンプル症候群(Temple syndrome, OMIM 616222)は、DLK1の発現低下(RTL1の発現低下も関与する可能性あり)によって主に引き起こされる【Kagami et al 2017b】。
テンプル症候群は、以下の遺伝的メカニズムによって発症する。
- 母親由来の第14染色体単親性ダイソミー(upd(14)mat)
- エピ変異(通常メチル化されている父親由来MEG3/DLK1:IG-DMRおよび父親由来MEG3:TSS-DMRの低メチル化)(父親由来の通常メチル化されたDMRが欠失しても、エピ遺伝的変化は引き起こされない。)
- DLK1(およびRTL1)の発現を阻害する父親由来インプリンティング領域の欠失や配列変異
テンプル症候群の臨床的特徴
- 筋緊張低下
- 出生前および出生後の成長障害
- 相対的な大頭症
- 乳児期の摂食困難
- 思春期早発症
鏡-緒方症候群との違い
- 胸郭および腹部の異常はテンプル症候群ではほとんど見られない【Prasasya et al 2020】。
鑑別診断
表3. 鏡-緒方症候群の鑑別診断として考慮すべき疾患
| 遺伝子/ 遺伝的メカニズム |
疾患 | 遺伝形式(MOI) | 鏡-緒方症候群と共通する特徴 | 鏡-緒方症候群と異なる特徴 |
|---|---|---|---|---|
| 11p15.5領域の異常メチル化、11p15.5を含むコピー数変異(CNV)、CDKN1Cの病的バリアント | ベックウィズ・ヴィーデマン症候群(Beckwith-Wiedemann syndrome) | 注釈.1参照 |
|
|
| DYNC2H1、IFT140、KIF7、NEK1、WDR19、WDR34を含む20以上の遺伝子 | 短肋胸郭異形成症(以前の名称:窒息性胸郭異形成症、ジューン症候群)(OMIM PS208500) | 常染色体劣性(AR) | - 小さな胸郭 |
|
| GPC3 | シンプソン・ゴラビ・ベーメル症候群1型(Simpson-Golabi-Behmel syndrome type 1) | X連鎖(XL) |
|
|
略語
- AR = 常染色体劣性(autosomal recessive)
- CNV = コピー数変異(copy number variant)
- MOI = 遺伝形式(mode of inheritance)
- XL = X連鎖(X-linked)
注釈
- ベックウィズ・ヴィーデマン症候群の遺伝的メカニズムを特定することは、再発リスクの評価に不可欠である。
- 家族のほとんどは再発リスクが1%未満であるが、特定の遺伝的メカニズムによっては再発リスクが50%に達する可能性がある(詳細はベックウィズ・ヴィーデマン症候群の項を参照)。
臨床的マネジメント
鏡-緒方症候群に関する診療ガイドラインはこれまでに公表されていない。そのため、以下の推奨事項は、本疾患の管理に関する著者らの経験に基づいている。
初回診断後の評価(Evaluations Following Initial Diagnosis)
鏡-緒方症候群と診断された患者の疾患の範囲と必要な対応を把握するために、以下の評価(表4参照)が推奨される。(これらの評価が診断に至る過程で未実施の場合)
表4. 鏡-緒方症候群:初回診断後の推奨評価
| 評価対象(システム/ 懸念事項) |
評価項目 | コメント |
|---|---|---|
| 発達 | 発達評価 | 粗大運動・微細運動スキル、適応能力、認知機能、言語評価を含む。 |
| 早期介入・特別支援教育の評価 | ||
| 神経系 | 神経学的評価 | |
| 呼吸器系 | 出生直後の呼吸評価 | 乳児期および幼児期に、上気道または下気道感染による呼吸不全を発症する例が多い。 |
| 筋骨格系 | 整形外科・リハビリテーション医学・PT・OTの評価 | 関節拘縮・後弯側弯症の評価 移動能力・ADL・補助装置の必要性評価 粗大運動スキル向上のためのPT、微細運動スキル向上のためのOTの必要性評価 |
| 消化器系 / 摂食 | 消化器科・栄養・摂食チームの評価 | 身長・体重の評価 誤嚥リスク・栄養状態の評価 嚥下障害や誤嚥リスクがある場合、胃ろう造設の評価を考慮 |
| 心疾患 | 心エコー検査 | |
| 腫瘍発生リスク | 腹部超音波検査(肝芽腫のスクリーニング) 血清α-フェトプロテイン(AFP)測定 |
可能であれば、AFP値の解釈に経験のある医師(腫瘍専門医、臨床遺伝専門医など)が評価すべき。 |
| 遺伝カウンセリング | 遺伝専門家によるカウンセリング | 鏡-緒方症候群の本質、遺伝形式(MOI)、医学的・個人的な意思決定への影響について説明。 |
| 家族支援・リソース | 臨床医・ケアチーム・家族支援団体による支援 | 家族および社会的状況を評価し、以下の必要性を判断
|
略語
- ADL = 日常生活動作(activities of daily living)
- MOI = 遺伝形式(mode of inheritance)
- OT = 作業療法(occupational therapy)
- PT = 理学療法(physical therapy)
- AFP = α-フェトプロテイン
注: 遺伝カウンセリングは、臨床遺伝専門医、認定遺伝カウンセラー、または遺伝看護専門認定看護師が担当することが推奨される。
治療(Treatment of Manifestations)
生活の質の向上、機能の最大化、合併症の軽減を目的として、支持療法が推奨される。
理想的には、各分野の専門家による多職種連携医療が必要となる(表5参照)。
表5. 鏡-緒方症候群:症状に対する治療
| 症状/懸念事項 | 治療 | 考慮事項/その他 |
|---|---|---|
| 発達遅滞 / 知的障害 / 神経行動学的問題 | 発達遅滞 / 知的障害の管理指針に従う。 | |
| 呼吸不全 | 出生直後から人工呼吸管理・酸素療法がほぼ必須。 | 気管切開が必要となることもある。 上気道・下気道感染の監視と治療が重要。 |
| 骨格異常 | 後弯側弯症の治療(整形外科に準ずる)。必要に応じて装具または手術を考慮。 | 関節拘縮の治療(リハビリテーション医学に準ずる)。 |
| 低体重 / 成長障害 | 経管栄養がほぼ必須。 | 必要に応じて胃ろう栄養を実施。 摂食訓練・リハビリテーションが推奨される。 嚥下障害の臨床的徴候がある場合、臨床的評価およびX線嚥下造影検査を低閾値で実施。 |
| 心疾患 | 循環器科による治療。 | 薬物療法を要する場合があるが、鏡-緒方症候群において心臓手術を受けた報告はない。 |
| 肝芽腫 | 標準治療(外科的切除および化学療法)。 | |
| 家族 / 地域支援 | 適切なソーシャルワーク支援を確保し、家族が地域のリソース、レスパイト(介護者支援)、サポートにアクセスできるようにする。 | 緩和ケアの介入や訪問看護の必要性を継続的に評価。 |
発達遅滞 / 知的障害の管理に関する課題
鏡-緒方症候群の詳細な臨床経過についての知見が限られているため、個々の患者に適した慎重な経過観察が推奨される。
運動機能障害(Motor Dysfunction)
粗大運動機能障害
- 理学療法(Physical Therapy, PT)が推奨される。
- 移動能力の最大化
- 後天的な整形外科的合併症(関節拘縮、側弯症、股関節脱臼など)のリスク低減
微細運動機能障害
- 作業療法(Occupational Therapy, OT)が推奨される。
- 微細運動スキルの障害による適応機能の低下に対応するため
- 例:摂食、整容、着替え、筆記などの日常動作(ADL)支援
口腔運動機能障害
- 各診察時に評価が必要。
- 以下の症状がある場合は、臨床的摂食評価およびX線嚥下造影検査を実施すべき。
- 食事中のむせや嘔吐反射
- 体重増加不良
- 頻回の呼吸器感染症
- 原因不明の摂食拒否
- 経口摂取が安全な場合
- 摂食訓練(通常は作業療法士または言語療法士による)を推奨。
- 摂食の協調運動や感覚過敏による摂食困難を改善するための訓練。
- 安全確保のために、とろみをつけたり冷却したりして食事を調整する。
- 摂食障害が重度の場合
- 経鼻胃管(NGチューブ)または胃ろう(Gチューブ)の適応を検討する。
コミュニケーションの問題(Communication Issues)
- 表出言語(話す能力)に困難がある患者では、代替コミュニケーション手段の評価を検討すべき。
神経行動学的 / 精神医学的問題(Neurobehavioral / Psychiatric Concerns)
発達小児科医との相談が、適切な行動管理戦略を検討する上で有用となる可能性がある。
経過観察(Surveillance)
既存の症状のモニタリング、支持療法への反応の評価、新たな症状の発現の確認を目的として、以下の評価が推奨される。表6. 鏡-緒方症候群:推奨される経過観察
| 評価対象(システム/懸念事項) | 評価項目 | 頻度 |
|---|---|---|
| 発達 | 発達の進行および教育的ニーズの評価 | 各診察時 |
| 呼吸器系 | 誤嚥や呼吸不全の兆候の監視 | 各診察時 |
| 骨格異常 | 後弯側弯症および関節拘縮の進行の監視 | 各診察時 |
| 摂食 | 栄養状態および経口摂取の安全性の評価 | 各診察時 |
| 消化器系 | 便秘の監視 | 各診察時 |
| 心疾患 | 心エコー検査 | 年1回 |
| 肝芽腫 | 腹部超音波検査 血清α-フェトプロテイン(AFP)測定 |
3か月ごと(3~4歳まで) 1,2 |
| 家族 / 地域支援 | 家族の支援ニーズの評価(ソーシャルワーク支援、緩和ケア、訪問看護、遺伝カウンセリングなど) | 各診察時 |
出典
- Sakaria et al [2021]
- 肝芽腫スクリーニングの方法は、ベックウィズ・ヴィーデマン症候群のガイドラインを参考にしている【Kalish et al 2017, Kalish et al 2024】。
避けるべき要因(Agents / Circumstances to Avoid)
- 呼吸器感染症への曝露を避けること。
- 特に乳児期には、呼吸器感染症による呼吸不全を発症するリスクが高い。
リスクのある家族の評価(Evaluation of Relatives at Risk)
- 遺伝カウンセリングに関する項を参照(遺伝的リスクを有する家族の検査に関する問題)。
研究中の治療(Therapies Under Investigation)
- 米国(ClinicalTrials.gov)および欧州(EU Clinical Trials Register)での臨床試験情報を検索することで、さまざまな疾患や病態に関する研究の情報を得ることができる。
- 注意:本疾患に特化した臨床試験が存在しない可能性がある。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
MECP2遺伝子-関連疾患は、X連鎖性の遺伝である。
患者家族のリスク
発端者の両親
鏡-緒方症候群の再発リスクは、発端者における母親由来RTL1asの発現低下を招く遺伝的メカニズムによって決まる。
本疾患の原因となる遺伝的メカニズムには以下が含まれる:
- 父親由来の第14染色体単親性ダイソミー(upd(14)pat)
- 母親由来のMEG3/DLK1:IG-DMRおよびMEG3:TSS-DMRのエピ変異(過剰メチル化)
- 母親由来のDMR(MEG3/DLK1:IG-DMRおよび/またはMEG3:TSS-DMR)の欠失(RTL1asを含む場合あり)
- 母親由来のMEG3プロモーター(MEG3:TSS-DMR)とRTL1asの構造的連続性を破壊する転座(または逆位)
ほとんどの患者では、遺伝的メカニズムは新生突然変異(de novo)として発生するため、同胞への再発リスクは増加しない。
しかし、まれに、母親由来の欠失など、同胞に対する再発リスクを高める遺伝的異常を有する親が存在することがある。
遺伝学的検査の推奨
MEG3/DLK1:IG-DMRおよびMEG3:TSS-DMRの過メチル化が確認された場合、upd(14)pat、エピ変異、DMR欠失を区別するために下記を実施する。
- 欠失解析(MS-MLPAによるメチル化解析と併用されていない場合は追加で実施)
- 親由来解析(第14染色体の多型DNAマーカーを用いる)
家族へのリスク(Risk to Family Members)
プロバンドの両親(Parents of a proband)
鏡-緒方症候群の患者の両親は、本疾患を発症しないが、以下のような発症リスクを高める遺伝的異常を有している可能性がある。
1. 母親由来のDMR欠失(MEG3/DLK1:IG-DMRおよび/またはMEG3:TSS-DMR、RTL1asを含む場合あり)
- 母親由来の欠失を持つ同胞では、鏡-緒方症候群の再発が報告されている。
【Kagami et al 2008, Kagami et al 2015, Beygo et al 2015, Luk et al 2017】
プロバンドの母親が欠失を有している場合、母は遺伝の経路によって以下のいずれかの疾患を発症する可能性がある。
- 父親由来のDLK1を含む欠失を受け継いだ場合 → テンプル症候群(Temple syndrome)
- 母親由来の欠失を受け継いだ場合 → 鏡-緒方症候群(理論上)(現在までに、鏡-緒方症候群の母親が自らの子に同疾患を伝達した例は報告されていない)。
2. 母親由来の転座(または逆位)
- MEG3プロモーター(MEG3:TSS-DMR)とRTL1asの間の構造的連続性を破壊する転座または逆位。
3. 両親いずれかのロバートソン転座
- 第14染色体を含むロバートソン転座またはi(14q)の存在。
再発リスクを明確にするため、両親の遺伝学的評価が推奨される。
- 具体的な検査方法は、プロバンドの遺伝的メカニズムに依存する(「同胞へのリスク」の項参照)。
プロバンドの同胞(Sibs of a proband)
プロバンドの同胞(兄弟姉妹)へのリスクは、プロバンドの遺伝的メカニズムと両親の遺伝的状態によって決まる。
- プロバンドがupd(14)patの場合
- プロバンドの核型分析を推奨(ロバートソン転座またはi(14q)染色体の有無を確認)【Ogata & Kagami 2016】
- プロバンドに転座またはi(14q)染色体が確認された場合、または染色体状態が不明な場合は、両親の核型分析を推奨。
- 両親の核型が正常な場合
- upd(14)patはde novo変異として発生したと考えられ、同胞の再発リスクは1%未満と推定される。
- いずれかの親にロバートソン転座またはi(14q)がある場合
- 同胞の再発リスクは増加する。
- ただし、ロバートソン転座を介したupd(14)patが報告されているにもかかわらず、同胞における再発例は確認されていない【Wang et al 2020】。
- エピ変異(過メチル化)の場合 母親由来のDMR(差異メチル化領域)のエピ変異(過メチル化)が認められる場合、
- プロバンド(発端者)において、新生突然変異(de novo)として発生したと推定される。
- そのため、同胞(兄弟姉妹)への再発リスクは1%未満と考えられる。
- プロバンドおよび両親の追加検査は、再発リスクの明確化のためには不要。
注意
- 過メチル化の根本的な遺伝的メカニズムは不明である。
- 多領域インプリンティング異常(MLID)を伴う鏡-緒方症候群の報告はない【Kagami et al 2017a】。
- 母親由来の14q32.2インプリンティング領域の欠失の場合
- プロバンドで検出された欠失が母親の白血球DNAで検出されない場合
- この欠失はプロバンドにおいてde novoで発生したと推定され、同胞の再発リスクは1%未満と考えられる。
- 注意:母親の白血球DNAの検査では、体細胞モザイクのすべてのケースを検出できない可能性がある。また、生殖細胞系列(ゴナド)のみの欠失は検出できない(現在のところ、14q32.2インプリンティング領域の欠失における生殖細胞モザイクの報告はない。)
- プロバンドの母親が欠失を有する場合
- 同胞の再発リスクは50%。
- 母親由来の欠失を持つ同胞において、鏡-緒方症候群の再発が報告されている【Kagami et al 2008, Kagami et al 2015, Beygo et al 2015, Luk et al 2016】。
- 注意:もし欠失がDLK1を含む場合、母親がその欠失を父親から受け継いでいる場合、母親はテンプル症候群(Temple syndrome)である可能性が高い。
プロバンドの母親に対する標的欠失解析が推奨される。
母親由来の転座または逆位(MEG3プロモーターとRTL1asの連続性破壊)の場合
- プロバンドの母親の核型が正常な場合
- 転座または逆位は、プロバンドにおいてde novoで発生したと推定される。
- 同胞の再発リスクは1%未満。
- プロバンドの母親が同じ転座または逆位を有する場合
- 同胞の再発リスクは増加する。
プロバンドの母親に対する核型分析が推奨される。
プロバンドの子孫(Offspring of a proband)
- 鏡-緒方症候群の患者は、現在のところ生殖年齢に達している例が少なく、また、これまでに繁殖(生殖)した患者の報告はない。
- しかし、鏡-緒方症候群の生殖機能は保たれていると推測されるため、以下の再発リスクに関する問題が考慮されるべきである。
1. プロバンドが14q32.2インプリンティング領域の欠失を有する場合
- 子孫は、プロバンドの性別および欠失の範囲によって、鏡-緒方症候群またはテンプル症候群のリスクを持つ。
- プロバンドが女性の場合 → 子孫が鏡-緒方症候群となるリスクは50%。
- プロバンドが男性であり、欠失がDLK1を含む場合 → 子孫がテンプル症候群となるリスクは50%。
2. プロバンドが単親性ダイソミー(upd)を引き起こす染色体異常を有する場合
- (例:ロバートソン転座またはi(14q)染色体)
- プロバンドの子孫は、以下のいずれかのリスクを持つ。
- upd(14)patを介した鏡-緒方症候群
- upd(14)matを介したテンプル症候群
- プロバンドの性別、および片親性ダイソミーの発生メカニズム(トリソミー救済、モノソミー救済など)によって決まる。
その他の家族への影響(Other family members)
- プロバンドの母親が14q32.2インプリンティング領域の欠失をヘテロ接合体として持つ場合
- 母親の同胞(兄弟姉妹)は、同じ欠失を持つリスクがある。
- プロバンドの母親が14q32.2を含む染色体異常(転座など)を有する場合
- 母親の同胞も同じ染色体異常を有するリスクがある。
遺伝カウンセリングに関連した問題
家族計画(Family Planning)
- 遺伝リスクの決定および出生前診断・着床前遺伝子診断(PGT)の利用可能性についての議論は、妊娠前の時点で行うことが最適とされる。
- 罹患者の親には、遺伝カウンセリングを提供することが適切である。
- (子孫への潜在的リスクおよび生殖の選択肢についての説明を含む。)
出生前診断および着床前遺伝子診断(Prenatal Testing and Preimplantation Genetic Testing)
陽性の家族歴がある場合(Positive Family History)
ゲノム変異(Genomic Variants)
- プロバンド(発端者)で第14染色体q32.2インプリンティング領域を含む欠失や転座が確認された場合、以下の方法による出生前診断が可能である。
- 絨毛検査または羊水検査で採取した胎児DNAを用いた検査
- 着床前遺伝子診断(PGT) 【Sasaki et al 2014, Sabria-Back et al 2022】
- プロバンドがupd(14)pat(父親由来の第14染色体単親性ダイソミー)による鏡-緒方症候群を発症し、両親の染色体解析が正常である場合、またはエピ変異(過メチル化)が原因である場合
- 再発リスクは1%未満と推定されるため、出生前診断や着床前遺伝子診断は推奨されない。
メチル化変化(Methylation Changes)
- 胎児DNAのメチル化検査(MEG3/DLK1:IG-DMRおよびMEG3:TSS-DMRの異常メチル化を評価する目的)は推奨されない。
- 羊水由来のDNAは胎児のメチル化状態を評価するための最も信頼性の高い組織と考えられているが、偽陰性の報告がある【Eggermann et al 2016】。
- エピ遺伝的変化に関する出生前診断の限界について、遺伝カウンセリングが推奨される【Eggermann et al 2016】。
出生前所見
- 出生前超音波検査で明らかな鏡-緒方症候群の所見が認められない場合でも、臨床症状の多様性や胎児超音波検査の限界を考慮すると、再発リスクが完全に除外されるわけではない。
- 鏡-緒方症候群のリスクがある新生児は、呼吸および摂食の状態を慎重に観察する必要がある。
- 臍帯ヘルニアがある場合、第2三半期以降に母体血清α-フェトプロテイン(AFP)濃度が上昇する可能性がある【Campbell & Copel 2018】。
家族歴がない場合(Negative Family History)
- 以下のような鏡-緒方症候群を示唆する臨床所見が胎児に認められた場合、出生前診断を考慮できる。
- 羊水過多
- 特徴的な顔貌
- コートハンガー状の肋骨を伴う小さな胸郭
- 臍帯ヘルニア
- このような状況で実施された出生前診断では、鏡-緒方症候群と一致する遺伝的異常が判明しており、以下の報告がある。
- モザイクupd(14)patが確認された症例【Suzumori et al 2010, Chen et al 2019, Igreja da Silva et al 2019, Molinet Coll et al 2021, Li et al 2021, Kuriki et al 2022】。
- 出生前診断の利用については、医療従事者や家族内で意見の相違がある場合がある。
- 多くの医療機関では、出生前診断の利用を個人の判断と考えているが、これらの問題について議論することは有益である。
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A. 鏡-緒方症候群に関連するOMIMエントリー
| OMIM番号 | OMIMエントリー名 |
|---|---|
| 608149 | KAGAMI-OGATA SYNDROME |
分子病態(Molecular Pathogenesis)
鏡-緒方症候群の主な原因は、RTL1アンチセンス(RTL1as)の機能喪失によるRTL1の過剰発現発現(約2.5倍~5倍)である。RTL1asは、RTL1の発現を抑制するトランス作用リプレッサーとして機能する【Kagami et al 2015, Ogata & Kagami 2016】。通常、母親由来の14 q32.2領域には、非メチル化MEG3/DLK1:IG-DMRおよび非メチル化MEG3:TSS-DMRが存在し、RTL1asの正常な発現を維持している。RTL1asが母親アレルから発現することで、父親アレル上のRTL1発現が抑制される(図4A参照)。
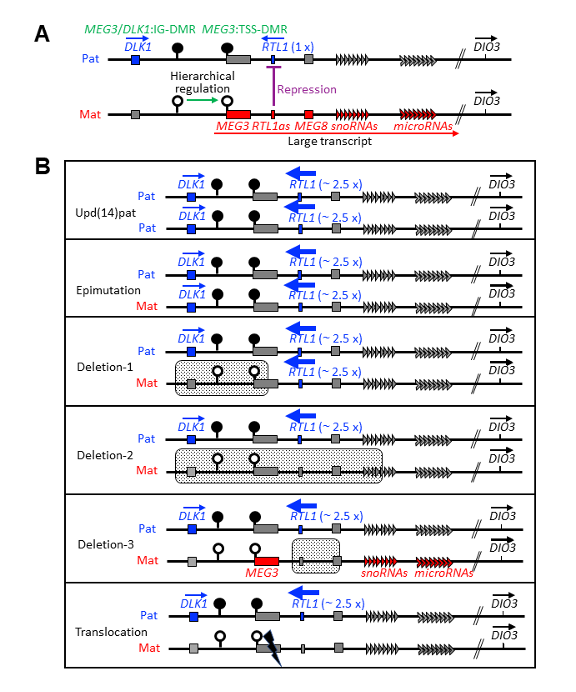
図 4.
染色体14q32.2のインプリンティング領域と、Kagami-Ogata症候群を引き起こす代表的な遺伝的原因。黒い円はメチル化されたDMR(差異的メチル化領域)を、白い円は非メチル化のDMRを示す。
A. インプリンティング領域の構造と特徴。
B. さまざまな状態におけるインプリンティング遺伝子の発現量。斑点部分は欠失領域を示し、はさみのマークは転座の切断点を示す。
第14染色体q32.2インプリンティング領域の構造と特徴
このインプリンティング領域には、以下の遺伝要素が含まれる(図4A参照)。
- 生殖細胞発生期由来のMEG3/DLK1:IG-DMR
- 受精後に形成されるMEG3:TSS-DMR)
- タンパク質コード遺伝子(父親アレルで発現):DLK1、RTL1
- 非コードRNA遺伝子(母親アレルで発現):MEG3、RTL1as、MEG8、snoRNA、miRNA
- 個体では、両DMR(MEG3/DLK1:IG-DMRおよびMEG3:TSS-DMR)は、父親由来の場合はメチル化され、母親由来の場合は非メチル化される。
- 胎盤では、MEG3/DLK1:IG-DMRはDMRとして維持されるが、MEG3:TSS-DMRは親由来に関わらず大部分が低メチル化される。
- MEG3:TSS-DMRとMEG3/DLK1:IG-DMRは、それぞれ個体および胎盤でインプリンティング制御センターとして機能する。
- MEG3:TSS-DMRの非メチル化は、MEG3/DLK1:IG-DMRが非メチル化されている場合にのみ維持される(両DMRのメチル化パターンは階層的に制御されている)【Kagami et al 2010】。
- すべての母親由来の非コードRNA遺伝子は、MEG3プロモーターによって制御される長い転写産物の一部である。
疾患発症のメカニズム(Mechanisms of Disease Causation)
鏡-緒方症候群の発症メカニズムには、以下が含まれる:
- 第14染色体父性片親性ダイソミー(upd(14)pat)
- 母親由来のMEG3/DLK1:IG-DMRおよびMEG3:TSS-DMRのエピ変異(過メチル化)
- 母親由来のMEG3/DLK1:IG-DMRおよび/またはMEG3:TSS-DMRの欠失
- 母親由来のRTL1アンチセンス(RTL1as)の欠失
- 母親由来のMEG3プロモーター(MEG3:TSS-DMR)とRTL1asの間の構造的連続性を破壊する転座または逆位
upd(14)pat(父親由来の第14染色体単親性ダイソミー)
- 高齢出産(母体年齢の上昇)は、モノソミー救済(monosomy rescue)を介したupd(14)patのリスク因子である【Kagami et al 2012a】。
エピ変異(過メチル化)
- 通常はメチル化されていない母親由来のMEG3/DLK1:IG-DMRおよびMEG3:TSS-DMRの過メチル化により、機能的なRTL1asが消失し、父親由来のメチル化パターンが形成される。
- 母親由来のMEG3/DLK1:IG-DMRの異常メチル化は、MEG3:TSS-DMRの異常メチル化を引き起こす。
- これまでに、MEG3/DLK1:IG-DMRまたはMEG3:TSS-DMR単独の過メチル化が報告された例はない。
- 過メチル化の根本的な原因は不明であり、鏡-緒方症候群の患者で多領域インプリンティング異常(MLID)は確認されていない【Kagami et al 2017a】。
DMRを含む欠失(Deletions Including Either DMRs)
- 母親由来のMEG3/DLK1:IG-DMRおよび/またはMEG3:TSS-DMRの欠失により、機能的なRTL1asが消失し、父親由来のメチル化パターンが形成される【Kagami et al 2010】。
- 母親由来のMEG3:TSS-DMR単独の欠失が3種類報告されており、いずれも父親由来のメチル化パターンを示した【Kagami et al 2010, Beygo et al 2015, Kilich et al 2024】。
- DMRの欠失がRTL1asを含む場合 → RTL1の発現量が通常の約2.5倍
- DMRの欠失がRTL1asを含まない場合 → RTL1の発現量が通常の約5倍
- 欠失がDLK1を含む場合 → DLK1の発現は正常
- 欠失がDLK1を含まない場合 → DLK1の発現量が通常の約2倍
- 母親由来の遺伝子(MEGs)は欠失または発現抑制される。
RTL1asの欠失(MEG3/DLK1:IG-DMRおよびMEG3:TSS-DMRを含まない場合)
- 母親由来の第14染色体q32.2領域におけるRTL1asの発現が消失し、その結果、RTL1の過剰発現が生じる。
- この場合、メチル化異常は認められない。
- これまで報告されたすべての例において、MEG8も欠失または破壊されている。
- DLK1および欠失されていない母親由来の遺伝子は正常に発現すると予測される。
転座によるMEG3プロモーターとRTL1asの破壊
- MEG3プロモーターとRTL1asの間の構造的連続性が破壊されることにより、RTL1asの転写が抑制される。
更新履歴
- 日本語訳者 :和田敬仁(信州大学医学部社会予防医学講座遺伝医学分野)
GeneReview 最終更新日: 2002.10.3. 日本語訳最終更新日 : 2003.8.21. (entried posting "Rett Syndrome") - GeneReview 著者 :Vicky L Brandt ;Huda YZoghbi , MD
.日本語訳者 :和田敬仁(信州大学医学部社会予防医学講座遺伝医学分野)
GeneReview 最終更新日: 2004.02.11. 日本語訳最終更新日 : 2006.1.18. (entried posting "Rett Syndrome") - Gene Review著者: John Christodoulou, MBBS, PhD, FRACP, FRCPA, FHGSA, Gladys Ho, BSc, MSc
日本語訳者: 伊藤雅之(国立精神神経センター神経研究所疾病研究第2部)、
久保田健夫(山梨大学大学院医学工学研究科環境遺伝医学講座)
Gene Review 最終更新日: 2009.4.2. 日本語訳最終更新日: 2010.3.3.( in present)
| X POST |
![]()