コフィン-ローリー症候群
(Coffin-Lowry Syndrome)
Gene Review著者:R Curtis Rogers, MD and Fatima E Abidi, PhD, DABMGG
日本語訳者: 槇野晋也、升野光雄、山内泰子、黒木良和 (川崎医療福祉大学大学院 医療福祉学研究科 遺伝カウンセリングコース)
Gene Review 最終更新日: 2018.2.1.日本語訳最終更新日: 2020.11.30
本項について:GeneReviewsではRPS6KA3関連知的障害 (RPS6KA3-RelatedIntellectualDisability)に統一された。
要約
疾患の特徴
コフィン-ローリー症候群(以下CLSと略す)は、通常、男性の重度から最重度の知的障害によって特徴付けられ、さほど重度ではない罹患者も報告されている。神経精神医学的懸念には、行動上の問題、筋力低下、進行性痙縮または対麻痺、睡眠時無呼吸、あるいは脳卒中が含まれる。刺激誘発転倒発作(SIDAs)(予期せぬ触覚や聴覚の刺激あるいは興奮が誘引となる意識喪失のない短時間の虚脱)が罹患者のおよそ20%にみられる。典型的なSIDAsは、幼児期中期と10代の間に始まる。特徴的な顔貌は、年齢とともにより明白になる場合がある。上肢の特徴はわずかなことがあり、肉付きが良い前腕だけでなく、手は短く、柔らかく、肉付きが良く、先細りの指を伴っている。進行性の脊柱後側弯症が、長期管理のうちで最も難しい側面のひとつである。罹患女性は、軽度から中等度の知的障害を有する傾向があり、男性でみられる典型的な顔貌、手、および骨格所見を示す場合もある。
診断・検査
CLSの診断は、ヘミ接合性のRPS6KA3病的変異を伴う男性において確立される。女性発端者におけるCLSの診断は、通常、分子遺伝学的検査により、ヘテロ接合性のRPS6KA3病的変異を伴う場合に確立される。ヘテロ接合性の女性において偏ったX染色体不活化以外の遺伝学的メカニズム(例:両アレルの RPS6KA3病的変異、または一方のX染色体にRPS6KA3病的変異をもつ女性で他方のX染色体の全体または部分的な欠失)が報告されている。罹患者の知的に正常な女性血縁者を注意深く診察することで、軽度の顔貌や手の症状が明らかになることがある。
臨床的マネジメント
症状の治療:
SIDAsは、クロナゼパムやバルプロ酸塩、選択的セロトニン再取り込み阻害薬あるいはベンゾジアゼピンのような薬剤によって治療されており、効果を改善するために、それぞれ投与量を至適化した異なる投薬試験が必要とされるだろう。頻繁にSIDAsを経験する者は、保護ヘルメットや車椅子の使用が求められるかもしれないし、可能ならば刺激するものから保護されるべきであろう。リスペリドンは、破壊的あるいは自傷行為がみられる人には有益かもしれない。哺乳困難、成長速度の異常、問題行動、脊柱後側弯症や肥満がもしみられたら、標準的な方法による治療を行う。
二次的合併症の予防:
心肺機能を危機にさらすまでの脊柱後側弯症の進行を妨げる介入。
経過観察:定期的な聴覚、歯、視覚の検査。毎年の臨床的な心臓の検査に加えて、5年から10年毎の心エコー図。進行性の脊柱後側弯症に対する脊柱の定期的な観察。
回避すべき薬品や環境:
SIDAsを経験している罹患者は、可能な限り驚かされることや転倒から保護されるべきである。
遺伝カウンセリング
CLSは、X連鎖形式で遺伝する。発端者のおよそ70%~80%はCLSの家族歴をもたず、20%~30%は、さらに1人以上の罹患した家族構成員をもつ。ヘテロ接合性であることが分かっている女性の子どもは、病的変異を受け継ぐリスクが50%である。病的変異を受け継いだ男性は罹患するだろう。病的変異を受け継いだ女性はヘテロ接合性となり、少なくとも何らかの発達遅滞と、軽いCLSの身体徴候の高いリスクがあるだろう。病的変異が罹患している家族構成員において確認されている場合には、リスクのある女性血縁者のための分子遺伝学的検査とリスクの増加した妊娠のための出生前検査が可能である。
診断
コフィン-ローリー症候群は、X連鎖の知的障害疾患である。この疾患が認められる人は典型的には男性であるが、女性罹患者も報告されている。この疾患に正式な診断基準はない。
示唆的な所見
次の所見がある男性では、コフィン-ローリー症候群(CLS)を疑う。
- 発達遅滞/知的障害.典型的な罹患男性は中等度から重度知的障害がある。分子遺伝学的検査の出現により軽度罹患男性が同定されるようになった [Field et al 2006]。
- 特徴的な頭蓋顔面像(特に罹患している年長児や成人において;図1,2と3参照)
- 通常、前額突出と濃い眉を伴う盛り上がった眼窩上縁
- 通常、眼瞼裂斜下を伴った際立った眼間開離、時折、軽い内眼角開離を伴った比較的正常な眼窩周囲域
- 一貫して、しばしば目立つ鼻の所見として、低い鼻梁、丸い鼻尖、厚みのある鼻翼と中隔、結果として生じる小さい鼻孔を含む
- 通常、開いたままの広い口、下口唇の反転した唇紅を伴った上下口唇の厚い唇紅
- 幼児期の粗な顔貌は、年齢と共に、より‘拳闘家’の容貌となる
- 突出した耳
- 上肢の特徴
- 短く、やわらかい、肉付きのよい手、しばしば目立って過伸展する指、小指球の一部を横切る短く水平な手掌線
- 比較的幅広い近位から遠位にかけて狭くなる際立った先細りの指、小さな末節骨と爪(図4参照)。手の特徴は時々微妙かもしれない(図5参照)。
- やわらかく、柔軟な手は、肥満者にみられるような、手のひらにほとんど‘フラシ天の座布団’のような感じを伴う
- 丸々とした、肉付きのよい前腕:年少児の診断において、もしかすると有用な徴候
- 筋骨格
- 頻繁に鳩胸や漏斗胸
- 幼児期に始まる脊柱後側弯症は、しばしば進行性である
注釈:何人かの著者は年少児における診断は難しいかもしれないと述べている。確かに、ほとんどの症候群においてよりも、CLSの顔の特徴は年齢と共にだんだん見分けられるようになる。しかしながら、新生児においてさえも良く考えれば、CLSの診断は、多くの場合明白である
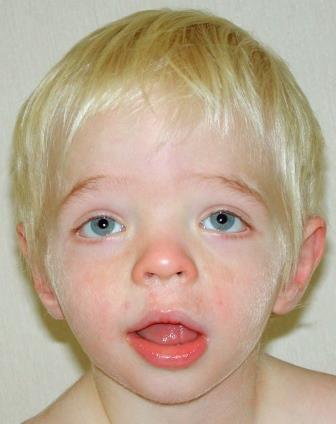 |
図1. CLSを伴った2歳男児の正面像。比較的軽微な顔貌特徴として、眼間開離、軽度の眼瞼裂斜下、幅広い鼻柱を伴った短い鼻、口唇の厚くわずかに反転した唇紅がみられる(患者は既知のRPS6KA3の病的変異をもつ)。 |
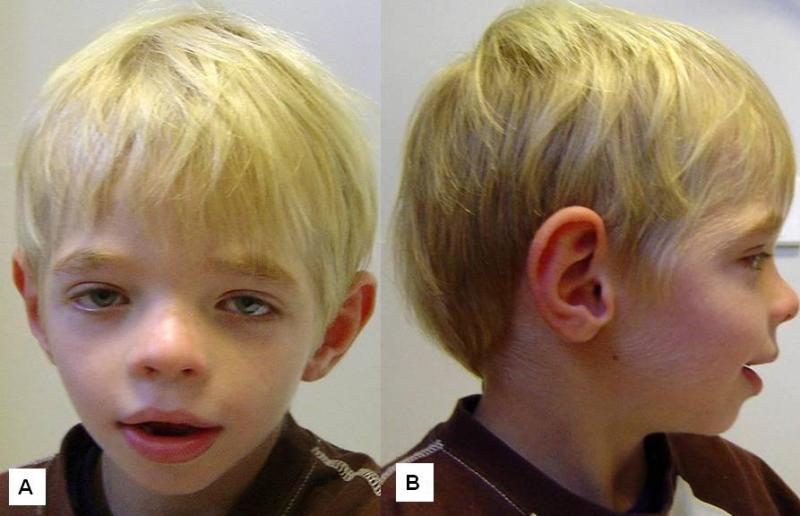 |
図2. 図1と同じ男児の5歳時の正面と側面像。より三角形の形をした粗な顔となり、典型的なCLSの顔貌の徴候の発現がみられる(患者は既知のRPS6KA3の病的変異をもつ)。 |
 |
図3. 青年の正面像。眼間開離、軽度の眼瞼裂斜下、上下口唇の厚い唇紅と小さい歯といった比較的軽度の顔貌の徴候がみられる。その鼻柱は幅広いが、鼻孔はちょうど良い大きさであり、疾患の発現における異人種間の違いが反映されているようである(患者は既知のRPS6KA3の病的変異をもつ)。 |
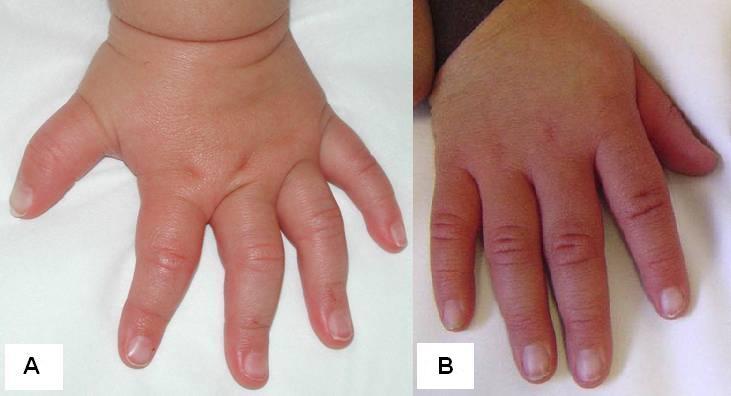 |
図4. 図1と2で例示した子供の2歳時(A)と5歳時(B)の手(患者は既知のRPS6KA3の病的変異をもつ)。 |
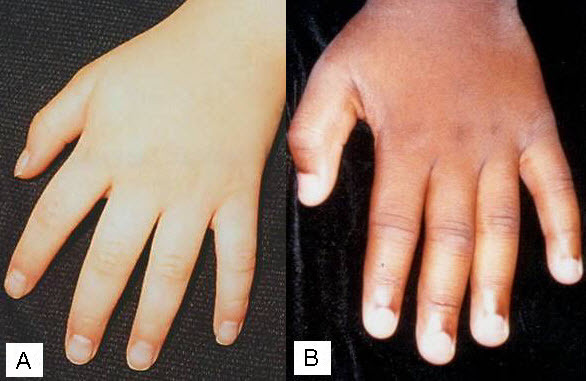 |
図5. A.古典的な先細りと柔らかい外見の年長児の手。 B.図3で例示した患者の手にみられた、よりわずかな特徴(患者は既知のRPS6KA3の病的変異をもつ)。 |
CLSのX線写真所見は、個々にはあるいはパターンとしては非特異的であるが、診断が疑われる際は有用な場合がある [Hanauer & Young 2002]。
- 大きな前頭洞を伴う厚い頭蓋
- 狭い椎間板腔と関連する変形性椎骨変化を伴う椎骨前縁突出
- 脊柱後側弯症
- 狭骨盤
- 中手骨の偽骨端、中節骨の過少モデリング、ドラムスティック状の末節骨(中手骨・指節骨プロフィールは診断の助けとはならないようである)。
- 一部の人では、頭部MRIで、軽度大脳萎縮、脳梁低形成、頭頂葉や前頭葉の脳室周囲白質病変、および/または大後頭孔の圧迫 [Tos et al 2015, Upadia et al 2017]
男性でCLSが疑われる特徴のいずれかを持つ女性ではCLSを疑うべきである。
- 完全な症状(特徴的な顔貌、手、骨格の所見を伴う軽度から中等度の知的障害を含む)をもつ
女性が報告されている [Young 1988,Plomp et al 1995,Fryssira et al 2002, Hunter 2002,Jurkiewicz et al 2010]
- 罹患者の知的に正常な女性血縁者の慎重な診察は、軽度の顔貌や手の症状発現を明らかにするかもしれない。
診断の確定
男性発端者. ヘミ接合性のRPS6KA3(RSK2としても知られている)病的変異を分子遺伝学的検査により同定することで男性発端者のCLSの診断が確立される(表1参照)。
女性発端者. ヘテロ接合性のRPS6KA3病的変異を分子遺伝学的検査により同定することで、通常は女性発端者のCLSの診断が確立される(表1参照)。ヘテロ接合性の女性において偏ったX染色体不活化以外の遺伝学的メカニズム(例:両アレルの RPS6KA3病的変異、または一方のX染色体にRPS6KA3病的変異をもつ女性で他方のX染色体の全体または部分的な欠失)が報告されている [Jacquot et al 2002]。
分子遺伝学的検査アプローチには、単一遺伝子検査、多遺伝子パネルの使用、およびより網羅的なゲノム検査が含まれる。
- 単一遺伝子検査.RPS6KA3シークエンス解析がはじめに行われ、病的変異が検出されない場合は標的遺伝子の欠失/重複検査が行われる。
- 多遺伝子パネル.RPS6KA3および関連のある他の遺伝子を含む多遺伝子パネル(鑑別診断を参照)が検討される場合がある。
注釈:(1)パネルに含まれる遺伝子や各遺伝子について用いられる検査の診断の感度は検査室によって異なり、時間経過とともに変わる可能性がある。(2)一部の多遺伝子パネルには、このGeneReviewで説明される疾患とは関連しない遺伝子が含まれる場合がある。したがって、臨床医は、一方で意義不明の変異や根本的な表現型を説明できない遺伝子の病的変異の同定を制限しつつ、最も手頃な費用で疾患の遺伝学的原因を特定できる可能性が最も高い多遺伝子パネルを決定する必要がある。(3)一部の検査室では、パネルオプションには臨床検査室が設計したカスタムパネル、あるいは臨床医が指定した遺伝子を含む表現型に特化したカスタムエクソーム解析が含まれる場合がある。(4)パネルで使用される解析方法には、シークエンス解析、欠失/重複解析、および/またはその他のシークエンスに基づかない検査が含まれる。
多遺伝子パネルの概要については、ここをクリック。遺伝学的検査を依頼する臨床医にとってより詳細な情報は、こちらをご覧ください。
- より網羅的ゲノム検査.(利用可能であれば)エクソームシークエンス解析やゲノムシークエンス解析を含むより網羅的ゲノム検査が考慮される。そのような検査は以前では考慮されなかった診断を提供するか示唆することもある(例えば、異なる遺伝子や類似の臨床所見を来す遺伝子の変異)。
網羅的ゲノム検査の概要については、ここをクリック。ゲノム検査を依頼する臨床医にとってより詳細な情報は、こちらをご覧ください。
表1.
コフィン-ローリー症候群に用いられる分子遺伝学的検査
| 遺伝子1 | 検査方法 | その方法により検出可能な病的変異2を有する発端者の割合 |
|---|---|---|
| RPS6KA3 | シークエンス解析3, 4, 5 | 90%〜95%6 |
| 標的遺伝子の欠失/重複解析7 | 5%〜10%6 | |
| 不明8 | 該当なし |
- 染色体座とタンパク質については表A.遺伝子とデータベース参照。
- この遺伝子で検出されたアレル変異についての情報は、分子遺伝学参照。
- シークエンス解析は、良性、おそらく良性、意義不明、おそらく病的、または病的変異を検出する。病的変異には、遺伝子内の小さな欠失/挿入、ミスセンス変異、ナンセンス変異、およびスプライス部位の変異が含まれる場合がある。通常、エクソンまたは全遺伝子の欠失/重複は検出されない。シークエンス解析結果の解釈で考慮すべき問題については、ここをクリック。
- シークエンス解析前のPCRによる遺伝子増幅の欠如は、罹患男性におけるX 染色体上の(多重)エクソンまたは全遺伝子の欠失を示唆する。確定には標的遺伝子欠失/重複解析による追加検査が必要となる。
- 注釈:Schneiderら[2013]は、通常のエクソン領域のシークエンス解析では検出されない異常なタンパク質をもたらす、深部イントロンの病的変異を特定した。
- ヒト遺伝子変異データベース(HGMD)、Delaunoy et al [2001]、Delaunoy et al [2006]
- 標的遺伝子欠失/重複解析は、遺伝子内の欠失または重複を検出する。用いられる手法には、定量PCR、ロングレンジPCR、MLPA法、および単一のエクソンの欠失/重複を検出するために設計された標的遺伝子マイクロアレイが含まれる。
臨床的特徴
臨床像
この疾患が認められる人は典型的には男性であるが、女性罹患者も報告されている。罹患女性は、男性でみられるような重度から非常に軽度までの所見を示す。男女ともに臨床診断が疑われる前に分子検査で、より多くの人が同定されるにつれ、より軽度の症状が認識されてきている。
Manouvrier-Hanu ら[1999]は、ミスセンス変異に関連した非常に軽度の症状を呈する同胞2例を報告した。著者らは、ファーストフードレストランで働くことができるRPS6KA3病的ミスセンス変異をもつ人を知っている [C Skinner私信]
診断時年齢は、身体的特徴や検査の利用可能性によって様々である。
発達
コフィン-ローリー症候群(CLS)は通常、男性の重度から最重度の知的障害を特徴とするが、軽度の障害をもつ人が報告されている [Young 1988, Hanauer&Young 2002, Hunter 2002, Pereira et al 2010]。罹患した女性は、軽度から中等度の範囲で知的障害を有する傾向がある。
- 早期の発達評価は、最終的な発達の予後を過大評価してしまうかもしれない [Hunter 2002]。早期の発達の遅れには幅があり、通常は運動発達よりも言語発達遅滞がより重度である。
- 筋緊張低下によって乳幼児期に摂食の問題を呈することがある。
神経精神医学
CLSをもつ人は、一般的に幸福で、のんびりしているようにしばしば記述されるが、自傷とその他の行動の問題が報告されている。
詳細な神経学的評価は、重度の知的障害により妨げられる可能性がある。報告された所見には以下が含まれる。
- 筋力と筋肉量の低下
- 深部腱反射の低下と亢進の両者
- 睡眠時無呼吸
- 脳卒中
- 進行性痙縮
- 歩行能力の喪失を伴った進行性の対麻痺(黄色靱帯の石灰化と先天性脊柱管狭窄症の両者によるものとみなされてきた [Hunter 2002])。
- 刺激誘発転倒発作(SIDAs)は、予期せぬ触覚や聴覚の刺激あるいは興奮が、下肢の60から80msの無筋電図活動を誘発した時に生じ、意識喪失がないにもかかわらず短時間の虚脱を招く [Crow et al 1998, Nakamura et al 1998, Hahn & Hanauer 2012]:
- SIDAsのビデオによる例証はNelson & Hahn [2003]参照
- 発症は4から17歳の間で、平均発症年齢は8.6歳である [Nakamura et al 2005]。
- SIDAs はCLS財団データベースでは20%(34/170人)に報告された [Stephenson et al 2005]。
年齢による変化.Stephenson ら[2005]は運動異常症の性質は年齢によって変化し、1個人は2種類以上の神経学的徴候をもつことがあることも強調した。症状発現の範囲は、刺激によって変化する情動脱力発作、過剰驚愕症、遷延性緊張性反応、および真のてんかん発作を含む。てんかん発作は、CLSをもつ人の約5%が冒される [Stephenson et al 2005]。
典型的な身体的特徴
示唆的な所見も参照
頭蓋顔面.特徴的な頭蓋顔面像(図1,2,3を参照)は、年齢とともに明らかになる場合がある。
上肢.症状の特徴はわずかなことがある。肉付きが良い前腕だけでなく、手は短く、柔らかく、肉付きが良く、先細りの指を伴っている。
筋骨格. 進行性の脊柱後側弯症は、CLSをもつ人の長期管理のうちで最も難しい側面のひとつである。正確な有病率は知られていないが、罹患男性の少なくとも47%と女性の32%が、進行性の脊柱後側弯症をもっていることを報告されている [Hunter 2002]。その割合は、整形外科の紹介クリニックからの報告においてより高かった [Herrera-Soto et al 2007]。公表された報告において、いかなる重篤さの公認の定義も採用されてはいないが、重篤さは、しばしば長い年月の間に進行し、脊柱後側弯症により呼吸器が危険にさらされることが早死にの一因となることは明らかである。
鳩胸あるいは漏斗胸の胸骨変形は頻度が高い。
X線写真にみられるその他の微小な骨格変化は、臨床的転帰をもたらさない。
心血管.罹患男性のおよそ14%と罹患女性の5%が、心血管疾患を有している [Hunter 2002]。CLSをもつ人の多くは、徹底した初回や継続した心臓の評価を受けていないので、これらの割合は、低く見積もられているだろう。報告は以下を含む:僧帽弁、三尖弁、大動脈弁の異常;短い腱索;心筋症(1人に心内膜線維弾性症を伴う);説明できない、うっ血性心不全;および大動脈と肺動脈の拡張 [reviewed in Hunter 2002]。Facherら[2004]により報告された1人に拘束性心筋症がみられた。Martinezら [2011]は、拘束性の左室心筋緻密化障害を伴うCLSをもつ人を報告した。心臓異常は、早死にの一因となるかもしれない。CLSをもつ人の心血管障害の系統的レビューはない。
成長.出生前の成長は正常である。成長障害は、通常、出生後の時期の早期に起こる。男性と重度の罹患女性は、一般的に身長の3パーセンタイルより下になるが、曲線をたどると予測される。低身長は、不均衡に短い下肢を反映しているかもしれない [Hunter 2002, Touraine et al 2002]。小頭症はよくあるが、多くのCLSをもつ人は、正常な頭囲をもつ。
歯科.歯の異常はよくあり、小さい歯、位置異常、開咬、永久歯の先天性欠如歯、乳歯の早期萌出、または萌出遅延、2つ以上の原因と思われる早期喪失を含む。口蓋は高い。年齢と共に年少児の下顎後退は、下顎前突に取って代わる傾向がある。
聴力損失.聴力損失は30%に報告されている [Pereira et al 2010]。Hunterは、14/89の罹患男性と1/22の罹患女性に聴力損失を報告した [Hunter 2002]。
- 聴力図は、感音性難聴を明らかにするだろう。
- 内耳奇形が後期発症の聴力損失の原因として報告されている [Rosanowski et al 1998]。
- 家族内における聴力損失の集積性が起こるかもしれない。
視覚の問題.重大な視覚の問題はめったにないようにみえるが、白内障、網膜色素の萎縮、視神経萎縮が報告されている。慢性的な瞼の炎症(眼瞼炎)の発生が増加しているかもしれない [reviewed in Hunter 2002]。
神経画像検査.脳の異常の一貫したパターンは示されていないが、以下の所見が報告されている [Tos et al 2015,Upadia et al 2017]。
- 菲薄化と無形成を含んだ脳梁の異常は何人かの著者によって報告されている [Kondoh et al 1998, Wang et al 2006]。
- MRIにて多発性限局性前頭葉の低信号 [Kondoh et al 1998]。
脳脊髄液の限局性領域によると考えられる低信号は、3人の罹患同胞においてWangら[2006]によって報告された。彼らは脳梁の菲薄化、小脳虫部の低形成と軽度の脳室の非対称も示した。著者らは、知的障害の程度は、MRI所見の重大さと相関すると結論を下した。
- 脳室周囲白質の異常
- 狭窄した大後頭孔(直径の縮小)
- 罹患男性と女性の定量的MRIにおいて、代償性脳室拡大を伴わない、灰白質と白質の容積低下は、細胞増殖の減少のような早期の神経発達異常を示唆する [Kesler et al 2007]。
神経病理学.脳回と層構造の異常は、剖検で指摘された [Coffin 2003]。
その他.一例のみに報告された所見は、直腸脱、子宮脱、空腸憩室、神経節細胞の減少を伴った結腸憩室、膝窩部のガングリオン、幽門狭窄、一側性腎無発生、前位肛門、顔の色素の増加と拡大した気管を含む [reviewed in Hunter 2002]。
死亡率.寿命は、CLSをもつ人の中には短縮することもある。文献に報告された人では、死亡は男性の13.5%と女性の4.5%で、平均年齢20.5(範囲:13-34)歳において起きていた [Hunter 2002]。
- 悪化させる要因は、心臓異常、汎細葉性肺気腫、呼吸器合併症、進行性脊柱後側弯症、痙攣に関連した誤嚥を含んでいた。
- Coffin [2003]は、彼の最初の患者の1人は、慢性的な肺と心疾患に肺炎を重ねた結果により18.8歳で死亡し、2番目の患者は、急性の食物誤嚥により18歳で死亡したと報告した。
- 著者らは、生命を脅かす中枢性と閉塞性の睡眠時無呼吸を呈したCLSをもつ人を知っており、顎の前進術の手術を行った後に呼吸器合併症により死亡した、慢性的な閉塞性と中枢性の睡眠時無呼吸の病歴をもつ別の男性を知っている。
- 1人の罹患した男性と1人の絶対ヘテロ接合性女性がホジキン病で死亡した [reviewed in Hunter 2002]。
ヘテロ接合性女性
ヘテロ接合性女性は、軽度の粗な顔貌で、先細りの指、低身長、正常な知能、またはさまざまな程度の知的障害を伴う著しく多様な表現型を示す。彼女らは、一般集団よりも高い割合で精神疾患を有するかもしれない。女性68人(CLS女性22人、非罹患のヘテロ接合体38人、‘罹患した’姉妹8人)中6人(8.8%)は、統合失調症、双極性障害、‘精神病’を含む精神疾患の診断を受けていた [reviewed in Hunter 2002]。Micheliら [2007]によって研究された2人の女性のうち1人が、‘精神病’を有していると記述され、Wangら[2006]によって2人の罹患している姉妹のうち1人が、統合失調症を有していると報告された。
遺伝型と表現型の関連
表現型とRPS6KA3病的変異の部位あるいは型との間に強い相関が存在する訳ではないが、一定の病的ミスセンス変異を伴った人は、より軽い疾患の表現をもつ傾向にある [Delaunoy et al 2001]。非症候群性知的障害の一つの型(MRX19;遺伝学的に関連する疾患を参照)をもつと分類された家族は、RPS6KA3にミスセンス変異をもっており、それはリボソームS6キナーゼ酵素活性の80%減少の原因となり、CLSをもつ人で、全てのリボソームS6キナーゼ酵素活性を失う原因となる多くの病的変異とは対照的である [Merienne et al 1999]。この所見は、いくつかのRPS6KA3変異は、おそらく非CLS表現型または非症候群性X連鎖知的障害を引き起こすことを示している。
Nakamuraら[2005]は、N末端キナーゼドメイン内か上流のどちらかで切断された変異は、刺激誘発転倒発作(SIDAs)特有の感受性の原因となるかもしれないと示唆した。しかし、タンパク質のC末端キナーゼドメインをコードする領域にヘテロ接合性病的変異を有するSIDAsをもつ罹患女性の所見は、この相関に反論するだろう [Rojnueangnit et al 2014]。
命名法
初期の著者らは、Lowryら[1971]によって報告された患者が同様の症候群であることが認められるまで、Coffin症候群と引用した。
いくつかの初期の教科書や論文は、Coffin-Siris症候群とCLSを混同していた。
有病率
CLSの推定有病率は公表されていない。著者らの経験に基づくと、1:40,000から1:50,000の割合が適当であろう。これは、しかしながら、実際の有病率を過小評価しているかもしれない。
遺伝学的に関連する疾患
非症候群性知的障害.RPS6KA3における病的変異に関連した非症候群性知的障害をもつ2家系がFieldsら[2006]によっても報告されている。
非症候群性知的障害の一つの型(MRX19)は、RPS6KA3の病的ミスセンス変異によることが示された [Merienne et al 1999]。病的ミスセンス変異と軽い知的障害のみを伴ったもう一人が、Delaunoyら[2001]によって報告された。
鑑別診断
年長男児や成人においてコフィン-ローリー症候群(CLS)の診断は、通常、問題を呈さない。年少児やより軽い罹患女性の所見は、他の症候群と重複するかもしれない。
- Borjeson-Forssman-Lehmann症候群(BFLS; OMIM 301900)は、重度の知的障害、CLSと似た手の所見、厚い中隔と小さい鼻孔を伴うこともある上向き鼻孔の短い鼻と脊柱後側弯症を特徴とするX連鎖劣性疾患である。追加の所見としては、大きく、突出した耳と視覚の問題である。BFLSをもつ人は、さらに極端な性腺機能低下症を呈し、著しい女性化乳房を呈する傾向にある。女性は、症候群の部分的な表現を示すかもしれない。欠けている所見は、著しい眼間開離、広い口と口唇の厚い唇紅である。PHF6遺伝子における病的変異が原因となる [Lower et al 2002]。
CLSは、Williams症候群、遺伝学的に異質なFG症候群、X連鎖アルファ-サラセミア知的障害(ATRX)症候群、およびPitt-Hopkins症候群といくつかの顔貌所見を共有するが、これらの疾患はいずれもCLSにみられる手の変化を示さず、各々が追加の区別する特徴を持つ:
- Williams症候群は、さらに心血管疾患(エラスチン動脈症、末梢肺動脈狭窄、大動脈弁上狭窄、高血圧)、結合組織の異常、知的障害(通常軽度)、特異的認知の側面、特有の性格特徴、成長障害と内分泌異常(高カルシウム血症、高カルシウム尿症、甲状腺機能低下症と思春期早発)を含む。哺乳困難は、しばしば乳児期において発育不全につながる。罹患者の99%以上は、エラスチン遺伝子(ELN)を含むWilliams-Beuren症候群の表現型の特徴に決定的な領域(WBSCR)の隣接した遺伝子欠失をもっている。Williams症候群は常染色体優性遺伝形式である。多くの症例は新生で起きる。
- FG症候群1型(MED12関連疾患を参照)は、X連鎖遺伝、知的障害、広い前額、眼瞼裂斜下を伴った眼間開離、下口唇の厚い唇紅、脊柱後側弯症、漏斗胸、特徴的な行動をCLSと共有する。不均衡性大頭、肛門の異常に関連する便秘、幅広い母指趾、突出した指尖の隆起、および小さく、丸い、しばしば折り重なった上部耳輪をもったカップ状耳介によって区別される [Graham et al 1998]。筋緊張低下は、しばしば関節制限に進展する。部分的な脳梁欠損と乳頭体の結合は比較的よくある。
- アルファサラセミアX連鎖知的障害(ATRX)症候群は、特有の頭蓋顔面像、性器異常、筋緊張低下を伴った重度の発達遅滞と知的障害、アルファサラセミアによる軽度から中等度の貧血によって特徴付けられる。性器異常は、尿道下裂と停留精巣から重度の尿道下裂と判別不明性器、46,XYの核型をもつ人における正常にみえる女性性器にまで及ぶ。ATRX症候群は、ATRX遺伝子の病的変異による。
- Pitt-Hopkins症候群は、年齢と共により明らかになる特有の顔貌、著しい発達遅滞/知的障害、および目覚めている間の一時的な過呼吸および/または息止めによって特徴付けられ、これは罹患者の約半数で起こる。他の良くみられる所見は、行動の問題、手の常同運動、痙攣、便秘、および重度の近視である。Pitt-Hopkins症候群はTCF4遺伝子の病的変異またはTCF4遺伝子が位置する染色体領域(18q21.2)の欠失に起因するTCF4遺伝子のハプロ不全による。
染色体異常症.様々な染色体異常症をもつ人がコフィン-ローリー症候群の特徴をもつことがある。2つの例:
- McCandlessら[2000]は、CLSを示唆する所見をもつ罹患者にdel(10)(q25.1q25.3)[10番染色体長腕中間部欠失]を伴った家族を報告した。
- Concannonら[2002]は、CLSの特徴と複雑な染色体再構成(2番,3番,7番および11番染色体を含む)をもつ人を報告した。
臨床的マネジメント
最初の診断後の評価
コフィン-ローリー症候群(CLS)と診断された人の疾患の程度とニーズを確かめるために、以下の評価が推奨される。
- 身長、体重、頭囲の計測
- 歩行、腸管あるいは膀胱機能の変化および、てんかん、あるいは運動異常症の評価のための病歴と神経学的診察
- 発達評価と介入計画の明確化
- 特に胸部と脊柱に注意した、徹底した筋骨格の診察、もし臨床的な適応があるときは、X線写真による評価
- 発達と年齢に応じた聴力の評価
- 歯科の評価
- 心臓の身体的診察と心電図、ベースラインとしての心エコー図
- 閉塞性睡眠時無呼吸を除外するためにポリソムノグラフィー検査
- 眼科評価、屈折と眼底鏡を含む
- 健康状態の徴候に対して適切な家族構成員の評価
- 子どものケアのための家族の能力の評価、特にもし母が知的に影響を受けている時には
- 臨床遺伝医および/または遺伝カウンセラーとの相談
症状の治療
CLSをもつ人は、コミュニケーションスキルの発達と、自立度を発達させるための活動とセルフケアへの参加といった、あらゆる機会を与えられるべきである。
刺激誘発転倒発作(SIDAs)を認識することは、刺激の引き金の発生を最小限にし、転倒からの保護を与える早期介入を可能にするだろう。
- 異なった投薬の試みと投与量を至適にする努力は、効果を改善するかもしれない [O'Riordan et al 2006, Arslan et al 2014]。
- クロナゼパムで治療された患者では、SIDAsが減少した [Arslan et al 2014]。
- 抗てんかん薬(AEDs)(例えば、バルプロ酸塩)、または選択的セロトニン再取り込み阻害薬の試みは、一般にそれらは効果がないが、適応となるかもしれない [Fryssira et al 2002]。
- ベンゾジアゼピンは、時々服用量の増加において、いくつかの症例において効果的であることを証明している [Touraine et al 2002, Nakamura et al 2005]。
- 様々な投薬によって改善されなかった1例において、Havaligiら[2007]は、ナトリウムオキシベートによる良い反応を報告した。
- もし頻繁に発作が起こるなら、保護ヘルメットの適応となり、車椅子の使用は、転倒と負傷を予防するために必要とされるだろう。
リスペリドンは、破壊的行為や自傷行為をあらわす人において有益かもしれない [Valdovinos et al 2002]。
哺乳困難、成長速度異常や肥満が存在するなら、標準的な方法で評価と治療をすべきである。
行動問題の治療は標準的なものであり、定期的な再評価を必要とする。
脊柱後側弯症の治療は標準的なものであるが、成人期に入るときに十分な再評価が必要である。
二次的合併症の予防
脊柱後側弯症と狭窄症のような背椎の問題の早期認識は、進行予防と生命を脅かすかもしれない長期における心血管系や神経系の合併症を防ぐための介入を可能にするだろう。
同様に、いくつかの心臓異常の早期認識は、二次的合併症の予防や適切な機能の延長を可能にするかもしれない。亜急性細菌性心内膜炎(SBE)の予防を必要とするCLSをもつ人もいる。
視覚と聴覚への注意は、いくつかの二次的な行動の変化を防ぐだろう。眼瞼炎の同定と治療は、眼をこすることと、起こりうる角膜の損傷を防ぐだろう。
歯の衛生と歯肉疾患への注意は、早期に歯を失うことのリスクを減少させるだろう。
経過観察
下記のものが適切である:
- 聴覚と視覚の定期的な検査
- 診断時の心臓評価および、その後の年1回の心臓の身体的診察。たとえ最初の心エコー図が正常でも、心筋症の発生率と発症年齢の範囲に関しての不確かさを考慮して、5~10年毎に行うべきである [Massin et al 1999, Facher et al 2004]。
- 進行性脊柱後側弯症の進展に対する脊柱の観察。歩行と排便/排尿習慣の変化、痛みの表出、およびクローヌスや腱反射異常のような局所の神経学的変化に注目した、脊柱管の狭小化を疑うすぐれた指標があるべきである。
- 一般集団におけるようなルーチンの歯科評価、しかし、歯の喪失のリスクへ特別の注意を払う。
注釈:CLSをもつ人のフォローアップのために提案されたガイドラインを含んだ表は、Hunter [2010]によって提供された。
回避すべき薬品や環境
SIDAsを経験したCLSをもつ人は、びっくりさせられることや転倒から可能な限り保護されるべきであ
る。
リスクにある血縁者の評価
遺伝カウンセリングの目的で、リスクにある血縁者を検査することに関する問題については、遺伝カウンセリングを参照。
研究中の治療法
広い範囲の疾患と健康状態に対する臨床研究情報にアクセスするために、米国のClinicalTrials.govおよび欧州のEU Clinical trails Registerを捜してみなさい。
注釈:この疾患における臨床試験はないかもしれない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
コフィン-ローリー症候群(CLS)はX連鎖遺伝の形式をとる。
発端者のおよそ70%~80%は、CLSの家族歴はなく、20%~30%は2人以上の罹患している家族構成員をもつ [Delaunoy et al 2001]。単一の事例(すなわち、1家族において1人のCLS)の高い発生率は、知的障害がみられるヘテロ接合の女性に対して生じる遺伝的選択に帰することができるかもしれない [Jacquot et al 1998a]。
患者家族のリスク
発端者の両親
- 男性
- 男性が家系内で唯一の罹患者(すなわち、単一の事例)である場合、母はヘテロ接合体であるか、罹患男性は新生のRPS6KA3病的変異をもつ可能性がある。後者の場合、母は保因者ではない。罹患男性のおよそ70%~80%は、単一の事例であり、おおよそ3分の2が新生で生じている [Pereira et al 2010]。
- 2人以上の罹患者を伴う家族においては、罹患男性の母は絶対ヘテロ接合体である。注釈:女性で罹患した子を複数持ち、他に罹患した血縁者がおらず、RPS6KA3病的変異が白血球DNAで検出できない場合、その女性は生殖細胞系列モザイクをもっている可能性が最も高い [Jacquot et al 1998b, Horn et al 2001]。
- 発端者の母は、粗な顔の特徴、厚い口唇、先細りの指のようなCLSの徴候の診察、および発端者で病的変異が同定されている場合には分子遺伝学的検査がされるべきである。
- 罹患男性の父親は、罹患者でも、RPS6KA3病的変異のヘミ接合でもないため、特に評価/検査を必要としない。
- 女性
- 女性の発端者は、母からRPS6KA3病的変異を受け継いでいるか、または理論的には父親の生殖細胞系列モザイクによるものだろう。
- 発端者の母は、CLSの徴候の診察、および発端者で病的変異が同定されている場合には分子遺伝学的検査がされるべきである。
- 両親の詳細な評価と広範な家族歴は、発端者が新生病的変異によるか、遺伝性の病的変異をもつかを区別するのに有用かもしれない。通常、母の分子遺伝学的検査により病的変異が遺伝性かどうかを判断できる。
発端者の同胞
- 発端者の同胞のリスクは、母の遺伝的状況に依存する。
もし発端者が女性の場合、理論的には父の遺伝的状況がRPS6KA3病的変異の生殖細胞系列モザイクの場合には、同胞のリスク要因となり得る。
- もし、発端者の母が病的変異をもっている場合、各々の妊娠において変異を伝える機会は50%である:
- 病的変異を受け継ぐ男性同胞は、罹患するであろう。病的変異を受け継ぐ女性同胞は、ヘテロ接合性となり、少なくともいくらかの発達遅滞とCLSの軽い身体的徴候を有する高いリスクがある。
- 無作為のX染色体不活化から予測されるように、軽い罹患女性が、重度に罹患した娘をもつかもしれない。
- ヘテロ接合の女性は、IQに相関しないX染色体不活化の軽度から中等度の偏りを示す [Simensen et al 2002]。
- 身体的徴候や知的障害の全くない、CLSの家族歴が知られていない発端者の母は、おそらくヘテロ接合であるリスクは低い。
- 生殖細胞系列モザイクは、この疾患において証明されている。このように、発端者に見出された病的変異が、母親のDNAにおいて確認されなかったとしても、発端者の同胞は、依然として、病的変異を受け継ぐリスクが増加している [Jacquot et al 1998b, Horn et al 2001]。
発端者の子
- CLSを伴った男性と重度に罹患している女性は、通常は生殖しない。
- CLSを伴った女性は、病的変異を各々の子供に伝える50%の機会をもっており、病的変異を受け継ぐ息子は罹患するであろう。娘はヘテロ接合性となり、少なくともいくらかの発達遅滞とCLSの軽い身体的徴候の高いリスクを有するであろう。
その他の発端者の家族
- もし、発端者の母が病的変異をもつことが発見されたなら、母の女性家族構成員はヘテロ接合のリスクがあるだろう(無症候性か症候性か)。そして母の男性家族構成員は、彼らの発端者との遺伝的関係に依存して、罹患リスクがあるだろう。
ヘテロ接合体(保因者)の検出
リスクにある女性血縁者が遺伝的状況を決定するための分子遺伝学的検査は、発端者において病的変異が確認されている場合に最も有用である。
注釈:(1)このX連鎖疾患におけるヘテロ接合の女性には、様々な臨床症状が現れる(臨床像を参照)。(2)女性のヘテロ接合体の同定には、(a)家族におけるRPS6KA3病的変異の事前同定が必要となるか(b)罹患男性が検査に応じられない場合は、分子遺伝学的検査を最初はシークエンス解析により、もし病的変異が確認できなかったら、標的遺伝子欠失/重複解析による。
遺伝カウンセリングに関連した問題
特定のカウンセリングの問題
- CLSを伴った発達遅滞の女性と彼女らの家族を生殖の選択と子供のケアに関して支援するために、相当な社会資源が必要とされるであろう。
- RPS6KA3病的変異が確認された、CLSの家族歴の知られていない発端者(すなわち単一の事例)の母の分子遺伝学的検査の結果の解釈には注意すべきである。生殖細胞系列モザイクが観察されている。従って、罹患した子孫において検出された病的変異が、母の白血球DNAに検出されないような時でさえ、出生前検査の提案は適切である。
家族計画
- 遺伝的リスクの決定、遺伝的状況の明確化、および出生前検査の有用性の討論のための最適な機会は妊娠前である。
- 罹患しているか、ヘテロ接合性であるか、またはヘテロ接合性であるリスクのある、若年成人女性に遺伝カウンセリング(子孫への潜在的なリスクと生殖の選択肢の討論を含む)を提供することは適切である。
DNAバンキング
DNAバンキングは、(通常は白血球から抽出された)DNAを将来の使用のために保存しておくものである。検査法並びに遺伝子、アレル変異および疾患に対する我々の理解が将来進歩するかもしれないので、罹患者のDNAの保存は考慮すべきである。
出生前検査および着床前遺伝学的診断
RPS6KA3病的変異が罹患した家族構成員で特定されると、リスクが高い妊娠の出生前検査と着床前遺伝学的診断が可能となる。
資源
GeneReviewsのスタッフは、この疾患のある人とその家族の利益のために、以下の疾患特異的団体か上部支援団体、および/または登録を選択した。GeneReviewsは、他の組織によって提供された情報については責任をもたない。選択基準の情報については、ここをクリック。
- Coffin-Lowry Syndrome Foundation
Phone: 425-427-0939
Email: CoffinLowry@gmail.com
CLSF
- National Institute of Neurological Disorders and Stroke (NINDS)
PO Box 5801
Bethesda MD 20824
Phone: 800-352-9424 (toll-free); 301-496-5751; 301-468-5981 (TTY)
Coffin Lowry Syndrome Information Page
分子遺伝学
分子遺伝学とOMIMの表における情報は、GeneReviewにおける他の場所の情報と異なっているかもしれない:表は、より最近の情報が含まれているかもしれない。
表A.コフィン-ローリー症候群:遺伝子とデータベース
| 遺伝子 | 染色体座 | タンパク質 | Locus-Specific Databases(座特異的データベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| RPS6KA3 | Xp22.12 | リボソームタンパクS6 キナーゼ アルファ-3 | RPS6KA3@LOVD | RPS6KA3 | RPS6KA3 |
データは、以下の標準的な参照文献を編集したものである:HGNCによる遺伝子記号; OMIMによる染色体座;UniProtによるタンパク質名。リンクが提供されているデータベース (Locus Specific, HGMD, ClinVar) の記述については、 ここをクリック。
表B. コフィン-ローリー症候群のOMIM登録(OMIM のすべてを参照のこと)
| 300075 | リボソームタンパクS6キナーゼ, 90‐KD, 3; RPS6KA3 |
| 303600 | コフィン-ローリー症候群;CLS |
分子学的病因論
CLSに関連する遺伝子、RPS6KA3(RSK2)は、Rasシグナル伝達カスケードの一員である成長因子調節性セリン/スレオニンキナーゼをコードしている。ヒトは、4つの密接に関連したRPS6KA(RSK)遺伝子をもっている。各々の遺伝子は、2つの同一ではないキナーゼ触媒ドメインをもっており、両者は最大限の活性のために必要とされる [Yntema et al 1999, Yang et al 2004]。RSKファミリーのメンバーは、増殖と分化のような細胞事象に関与する。
遺伝子構造.RPS6KA3の転写物(NM_004586.2)は22個のエクソンを含み、これは、リボソームS6キナーゼ(別名:RSK2)にちなんで命名されている。遺伝子とタンパク質の情報の詳細な概要については表A遺伝子を参照。
病的変異.RPS6KA3における病的変異は、特定の表現型に関連した集積性の根拠はなく、遺伝子の至る所に分布している。
今までに最も大きな研究において(250人)、71の病的変異が86の血縁のない家族において発見された。ほぼ60%がタンパク質の短縮の原因となるか、または短縮が予測された。38%はミスセンス変異、20%はナンセンス変異、18%はスプライシングの異常、そして21% は遺伝子内の欠失か挿入であった [Delaunoy et al 2001]。
CLSが疑われた血縁のない106人のより小さな研究は、28の病的変異(26%)を見いだした。28の病的変異のうち、60%はタンパク質の短縮の原因となるか、または短縮が予測された。36%はミスセンス変異、21%はナンセンス変異、11%はスプライシングの異常、そして32%は遺伝子内の欠失か挿入であった [Abidi & Schwartz, 未発表]。
スプライシング部位の病的変異とタンパク質の正常機能を崩壊させるLINE‐1配列のイントロンへの挿入が報告されている [Zeniou et al 2002, Martinez-Garay et al 2003, Zeniou et al 2004]。(より多くの情報には、表A参照)
最近、Schneiderら[2013]は、異常なタンパク質をもたらす深部イントロンの病的変異を特定した。この発見は、CLSの臨床診断が非常に疑われ、エクソン解析で病的変異が検出できなかった全ての人に対してRNAレベルでの病的変異解析を正当とする。
全体あるいは部分的な遺伝子重複が報告されている。Matsumotoら [2013]は、軽度知的障害、注意欠陥多動障害、局在性てんかん、広汎性発達障害(PDD)をもつ家族におけるRPS6KA3遺伝子全体を含む微小重複を報告している。Marques Pereira ら[2007]は、CLSをもつ人にインフレームの縦列多重エクソン重複を報告している。遺伝子内のAlu配列の頻度が高いことを考慮すると、これらが比較的一般的な出来事であることを示唆している。ただし、さらなる研究が必要である。
正常遺伝子産物.リボゾームタンパクS6キナーゼアルファ-3(RPS6KA3)は、ras-MAPK、プロテインキナーゼC、アデニルシクラーゼを含む多くの経路でキナーゼ活性化に関与している [Harum et al 2001, Pereira et al 2010]。RPS6KA3は神経突起形成を調節し [Ammar et al 2013]、開口分泌に必要な脂質を生成するためにPLD1の活性化を仲介して [Zeniou-Meyer et al 2008, Zeniou-Meyer et al 2009]、神経伝達物質の放出を調節する [Zeniou-Meyer et al 2010]。
CLSだけでなく、非症候群性のXLMR(MRX19;遺伝学的に関連する疾患を参照)とRPS6KA3との関連は、その遺伝子は認知機能に決定的に重要であることを示している。
RPS6KA3の発現は、ヒトの胚形成において、時間的な制約と空間的な制約の両者を示し、妊娠9週の終脳から菱脳にかけて均一に脳に発現がみられる [Guimiot et al 2004]。RPS6KA3は、さらにCREB(サイクリックAMP応答配列結合タンパク質)を活性化させることが示され、それは、神経細胞の生存と、短期記憶から長期記憶への転換に関係している [Harum et al 2001]。
RPS6KA3は、細胞周期の進行とDNA修復を仲介することにより、ゲノムの安定性を維持する上でも重要な役割を果たす [Lim et al 2013]。MAPK/RSK経路および上皮細胞成長因子(EGF)によるヒストン H3のリン酸化により、G0とG1の間の細胞周期の刺激に役割を果たすようである。
異常遺伝子産物.RPS6KA3遺伝子における病的変異は、CLSと非症候群性のXLMRの両方の原因となる。CLSをもつ人における病的変異は遺伝子産物のキナーゼ活性の喪失を生じる。しかしながら、MRX19に関連した病的変異は、キナーゼドメインの外部で起こり、RPS6KA3活性の減少を生じる。それは、脳は、CLSで冒されるその他の臓器系よりも RPS6KA3活性の水準により敏感であることを示している。
Harumら[2001]は、7人の被験者で、IQとリンパ芽球におけるRPS6KA3を介したCREBtideリン酸化反応の減衰の程度との相関を示した。
Yangら[2004]は、RPS6KA3によるATF4のリン酸化の欠如が骨芽細胞の分化におけるATF4の正常な調節の役割を妨害しており、それがCLSで見られる骨の異常の一部を説明でき、おそらく脊柱後側弯症の進行性についても同様に説明できることを示唆している。
更新履歴:
- Gene Review著者:Alasdair GW Hunter, MD, Fatima E Abidi, PhD
日本語訳者:市川真臣、山内泰子、升野光雄、黒木良和
(川崎医療福祉大学大学院医療福祉学研究科遺伝カウンセリングコース)
Gene Review 最終更新日: 2009.1.15 日本語訳最終更新日:2010.5.17. - Gene Review著者:R Curtis Rogers, MD and Fatima E Abidi, PhD, DABMGG
日本語訳者: 槇野晋也、升野光雄、山内泰子、黒木良和 (川崎医療福祉大学大学院 医療福祉学研究科 遺伝カウンセリングコース)
Gene Review 最終更新日: 2018.2.1.日本語訳最終更新日: 2020.11.30[in present]
![]()