Baller-Gerold症候群(バレーゲロルド症候群)
(Baller-Gerold Syndrome)
Gene Reviews著者: Lionel Van Maldergem, MD, PhD, Juliette Piard, MD, Lidia Larizza, MD, and Lisa L Wang, MD.
日本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2018.4.19. 日本語訳最終更新日: 2023.5.28.
要約
疾患の特徴
Baller-Gerold症候群(BGS)は、出生時、児に頭蓋縫合早期癒合と上肢奇形が認められる場合に疑われることのある疾患である。癒合が生じるのは冠状縫合であることが最も多いものの、前頭縫合、ラムダ縫合、矢状縫合についても、単独で、あるいは組み合わさった形で癒合が生じることがある。上肢の異常としては、拇指の低形成/無形成や橈骨の低形成/無形成が、時に非対称な形で組み合わさって認められることがある。手根骨や中手骨の奇形や無形成の報告もみられる。皮膚の病変は、生後の数年間のどの時期にも現れうる。通常、まず顔や四肢の紅斑に始まり、多形皮膚萎縮へと変化していく。乳児期の段階から明らかな発育遅延がみられ、最終身長は、通常、平均値に対し-4SDのレベルとなる。
診断・検査
発端者におけるBGSの診断は、これに特徴的な臨床症候を有すること、ないしは、分子遺伝学的検査にてRECQL4の両アレル性病的バリアントが同定されることをもって確定する。
臨床的マネジメント
症状に対する治療:
両側性の頭蓋縫合早期癒合に対しては、生後6ヵ月未満の段階で修復術を行う。物を掴む機能を作り出すため、示指に対して拇指化手術を行う。多形皮膚萎縮については、皮膚癌に関する防護のため、日焼け止めを使用する。
定期的追跡評価:
RECQL4関連の諸疾患を有する罹患者は、骨肉腫やリンパ腫に関し高リスクであるため、BGS罹患者については、骨肉腫の臨床所見(例えば、骨痛、腫脹、跛行)、ならびに、リンパ節腫脹やリンパ腫の全身症候(発熱や、原因不明の体重減少)などの臨床所見に注意を払うことが推奨される。
避けるべき薬剤/環境:
皮膚癌のリスクがあるため、日光への曝露を避ける。
遺伝カウンセリング
Baller-Gerold症候群は常染色体潜性の遺伝形式をとる。罹患児の両親は、病的バリアントを1つ有する絶対ヘテロ接合者である。ヘテロ接合者(保因者)は無症候である。罹患者の同胞は、受胎の段階で、罹患者である可能性が25%、無症候の保因者である可能性が50%、罹患者でも保因者でもない可能性が25%である。家系内に存在する病的バリアントが2つとも同定されている場合には、高リスクの妊娠に備えた保因者診断のための検査や、着床前遺伝学的検査を行うことが可能である。
診断
本疾患を示唆する所見
次のような所見を組み合わさった形で有する例については、Baller-Gerold症候群を疑う必要がある。
- 冠状縫合の早期癒合
これは、臨床的には、眼球突出と目立つ額を伴う頭蓋の形の異常(短頭症)という形で現れる。これは、頭蓋骨のX線写真、あるいは、より好ましくは3D-CTの再構成で確認することができる。
冠状縫合が癒合すると、眼窩は前方へ引っ張られる。なお、冠状縫合は、前方からの投影では確認できない。これは、ラムダ縫合も同じである。
- 橈側列の奇形
これは、拇指の無形成あるいは低形成、ないし、橈骨の無形成あるいは低形成という形で現れる。
注:橈側列の軽度の奇形については、確認にX線写真が必要になる場合がある。
- 発育不全
- 多形皮膚萎縮
多形皮膚萎縮は、点状萎縮と毛細血管拡張を伴う皮膚の高色素沈着と低色素沈着という形で現れる。
本疾患を示唆する所見
次のような所見を組み合わさった形で有する例については、Baller-Gerold症候群を疑う必要がある。
- 冠状縫合の早期癒合
これは、臨床的には、眼球突出と目立つ額を伴う頭蓋の形の異常(短頭症)という形で現れる。これは、頭蓋骨のX線写真、あるいは、より好ましくは3D-CTの再構成で確認することができる。
冠状縫合が癒合すると、眼窩は前方へ引っ張られる。なお、冠状縫合は、前方からの投影では確認できない。これは、ラムダ縫合も同じである。
- 橈側列の奇形
これは、拇指の無形成あるいは低形成、ないし、橈骨の無形成あるいは低形成という形で現れる。
注:橈側列の軽度の奇形については、確認にX線写真が必要になる場合がある。
- 発育不全
- 多形皮膚萎縮
多形皮膚萎縮は、点状萎縮と毛細血管拡張を伴う皮膚の高色素沈着と低色素沈着という形で現れる。
方法1
表現型と臨床検査所見からBaller-Gerold症候群が示唆されるようであれば、使用する分子遺伝学的検査のアプローチは、単一遺伝子検査、あるいはマルチ遺伝子パネルといったものになろう。
- 単一遺伝子検査
RECQL4の配列解析を行うことで、遺伝子内の小欠失/重複やミスセンス・ナンセンス・スプライス部位バリアントが検出されるが、通常、エクソン単位、あるいは遺伝子全体の欠失/重複といったものは検出されない。
順番としては、配列解析を最初に行う。そこで病的バリアントが1つも、あるいは1つしか検出されない場合は、次いで遺伝子内の欠失や重複を検出するための遺伝子標的型欠失/重複解析を行う。
- マルチ遺伝子パネル
現況の表現型と直接関係のない遺伝子の意義不明バリアントや病的バリアントの検出を抑えつつ、疾患の遺伝的原因の特定に最もつながりやすいのは、RECQL4その他の関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネルであるように思われる。
注:(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネルの配列解析で病的バリアントが1つしか、あるいは1つも検出されない場合は、欠失/重複解析も含んだマルチ遺伝子パネルを検討する必要がある(表1参照)。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝学的検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
方法2
表現型からは他に数多く存在する遺伝性の頭蓋縫合早期癒合を呈する疾患と区別ができない例、あるいは、表現型が非典型的なものであるためBaller-Gerold症候群を頭に入れるところにまでは至らないといった例については、網羅的ゲノム検査(この場合は臨床医の側で疑わしい遺伝子の目星をつけておく必要はない)が最良の選択肢となろう。エクソームシーケンシングが最も広く用いられているが、ゲノムシーケンシングを使用することも可能である。
エクソームシーケンシングで診断がつかなかった場合は、もし臨床的に利用可能なようなら、エクソームアレイも検討対象になりうる。
網羅的ゲノム検査の基礎的情報についてはここをクリック。
ゲノム検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
表1:Baller-Gerold症候群で用いられる分子遺伝学的検査
| 遺伝子1 | 方法 | その手法で病的バリアント2が検出される割合 |
|---|---|---|
| RECQL4 | 配列解析3 | 95%超4 |
| 遺伝子標的型欠失/重複解析5 | 稀6,7 |
- 染色体上 の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- アレルバリアントの情報については、「分子遺伝学」の項を参照のこと。
- 配列解析を行うことで、benign、likelybenign、意義不明、likelypathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、遺伝子内の小欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。 配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
- Larizzaら[2013]
- 遺伝子標的型欠失/重複解析では、遺伝子内の欠失や重複が検出される。具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失/重複の検出を目的に設計された遺伝子標的型マイクロアレイなど、さまざまなものがある。
- 遺伝子標的型欠失/重複解析の検出率に関するデータは得られていない。
- 日本人起源のBaller-Gerold症候群の1例で、遺伝子内の大欠失のホモ接合が報告されている[Kanekoら2017]。
臨床的特徴
臨床像
Baller[1950]とGerold[1959]が最初にBaller-Gerold症候群(BGS)を報告して以降、これまでに40人弱のBGS罹患者が報告されている[Mégarbanéら2000,VanMaldergemら2006,Debeljakら2009,Siitonenら2009,Piardら2015,Kanekoら2017]。BGSは、出生の段階で児に頭蓋縫合早期癒合と上肢の異常がみられる場合に、疑われることのある疾患である。早期癒合は、冠状縫合に最も多く現れるものの、前頭縫合、ラムダ縫合、矢状縫合に、単独で、あるいは組み合わさった形で癒合が生じることもある[VanMaldergemら2016]。
頭蓋縫合早期癒合に伴ってみられる頭蓋顔面所見
- 短頭症
- 眼球突出
- 突出した前額
- 大きな大泉門
その他の頭蓋顔面症候
- 凹んだ鼻梁
- 短い鼻
- 赤唇の薄い小さな口
- 高口蓋
骨格奇形
- 上肢の奇形
拇指の低形成/無形成、橈骨の低形成/無形成が、組み合わさった形でみられるが、現れ方は時に非対称なこともある。手根骨や中手骨の奇形あるいは欠損の報告もみられる。
- 膝の異常
膝蓋骨の低形成/無形成は、小児期に明らかになる。
- 膝蓋骨の骨化遅延は、乳児期において、膝蓋骨の欠損と誤って認識されることがある。
- 膝蓋骨が欠損すると、反張膝や膝の不安定性の形で現れることがある。
皮膚所見
皮膚の病変は、出生後の数年間については、いつでも現れる可能性がある。
- 通常、病変は、最初、顔や四肢の紅斑の形で現れる。
- これは、その後、多形皮膚萎縮(斑状の低色素沈着と高色素沈着,萎縮,毛細血管拡張)へと変化していく。
成長
乳児期の段階から明らかな発育遅延がみられ、最終身長は、通常、平均値に対し-4SDのレベルとなる。
発達/知能
知的障害の報告がみられる[RamosFuentesら1994]。それでも、全例とまでは言えないまでも大多数の罹患者については、知能は正常である。知的発達に関する正式な研究は、これまで行われていない。
他の所見
- 数例で、鎖肛あるいは前方肛門が報告されている。
- 時に、心室中隔欠損、Fallot四徴、先天性門脈奇形などの心血管異常の報告がみられる。
がんのリスク
Baller-Gerold症候群の1例でリンパ腫が報告されている[Debeljakら2009]。ただ、RECQL4の病的バリアントに起因して生じるその他の疾患(「遺伝子の上で関連のある疾患」の項を参照)においては、骨肉腫、リンパ腫、皮膚癌に関し高リスクであることが報告されている。そのため、がんを示唆する症状を有するBaller-Gerold症候群罹患者は、迅速に評価を受ける必要がある
遺伝型-表現型相関
これまでに報告された罹患者の数が限られていることから、今のところ正式な形で確立した遺伝型-表現型相関は存在しない。
疾患名について
Baller-Gerold症候群という名称は、BallerならびにGeroldによる3人の罹患者を報告したドイツの文献をもとに、Cohen[1975]が命名したものである。
- Baller[1950]は、低身長、尖頭、左橈骨の低形成、右橈骨の無形成を呈する1女性を報告している。この女性の両親は遠縁関係にあった。
- Gerold[1959]は、頭蓋縫合早期癒合、橈骨と拇指の無形成、尺骨の彎曲を有する男女の同胞例を報告している。
1975年以降、あらゆるタイプの橈側列の奇形を伴うあらゆるタイプの頭蓋縫合早期癒合に対して、Baller-Gerold症候群という名称が用いられてきた。しかし、これは正しくない使用法であるように思われ、一部に、バルプロ酸胎芽病でみられる前頭隆起と橈側列の奇形についてもBGSの診断基準を満たすものであると考える向きもある[SantosdeOliveiraら2006]。
発生頻度
Baller-Gerold症候群の発生頻度はよくわかっていない。おそらく、1,000,000人に1人を下回るものと思われる[Moら2018]。
遺伝学的に関連のある疾患(同一アレル疾患)
RECQL4の病的バリアントは、Rothmund-Thomson症候群罹患者[Kitaoら1999]やRAPADILINO症候群罹患者[Siitonenら2003]においても同定されている。
- Rothmund-Thomson症候群(RTS)
Rothmund-Thomson症候群は、多形皮膚萎縮、疎な毛髪・睫毛・眉毛、低身長、骨格と歯の異常、白内障、発がん(特に骨肉腫)リスクの上昇を特徴とする疾患である。皮膚は、通常、出生時は正常で、生後3-6ヵ月に紅斑・腫脹・水疱の形で顔面に疹が現れ、その後、臀部や四肢にそれが広がる。疹は、数ヵ月から数年をかけて、慢性の網状低色素・高色素沈着、点状萎縮、毛細血管拡張(これらは、ひとまとめにして多形皮膚萎縮の名で知られる)に移行する。過角化性病変がおおむね3分の1の罹患者にみられる。骨格異常としては、橈側列の奇形、尺骨の奇形、膝蓋骨の無形成あるいは低形成、骨減少症などがある。
RTSの診断は、臨床所見に基づいて行われる。分子レベルの検査は確認用であり、臨床所見が非定型的であるような場合にこれが有用な場合がある。臨床所見で確定しにくいような場合は、分子遺伝学的検査でRECQL4の両アレル性病的バリアントが同定されることで、診断が確定する。
- RAPADILINO症候群(OMIM266280)
RAPADILINOというのは、radialraydefect(橈側列の奇形)、patellahypoplasiaoraplasia(膝蓋骨の低形成あるいは無形成)、cleftorhighlyarchedpalate(口蓋裂あるいは高口蓋)、diarrheaanddislocatedjoints(下痢と関節脱臼)、littlesizeandlimbmalformation(低身長と四肢奇形)noseslenderandnormalintelligence(細い鼻,正常知能)の頭字語である。この疾患は、出生前、出生後の発育遅延を特徴とする。頸椎分節障害が報告されている。摂食障害と原因不明の若年性下痢に起因して、成長障害が生じる[Siitonenら2003]。フィンランドで最初に報告されて以降[Kääriäinenら1989]、これまでに14人のフィンランド人、2人のフィンランド人以外の人が報告されているのみである[Vargasら1992,Kantら1998,Jamら1999,Siitonenら2003]。16人中1人に骨肉腫が報告されている。RAPADILINO症候群罹患者では、リンパ腫がしばしばみられる合併症であるように思われ、35歳未満の4例にこれがみられている[Siitonenら2009]。
RAPADILINO症候群を引き起こすRECQL4のフィンランド人特異的スプライス部位バリアント(IVS7+2delT)では、エクソン7のインフレームスキッピングが生じ、保存された領域であるヘリカーゼドメイン直前の44のアミノ酸が取り除かれるだけで、見かけ上、ヘリカーゼドメインそのものの転写には変化が生じないと予測される。14人のフィンランド人罹患者中9人は、IVS7+2delTのホモ接合、5人は、IVS7+2delTとヘリカーゼ外であるエクソン5、18、19のナンセンスバリアントとの複合ヘテロ接合である。すなわち、ヘリカーゼドメインはどの例についても損なわれずに残っているということであり、多形皮膚萎縮や、骨肉腫への素因に関して、こうした点が一定の役割を果たしているものと考えられている[Siitonenら2003]。
RTS、RAPADILINO症候群、BGSは、出生前・出生後の発育遅延、慢性下痢、膝蓋骨の低形成あるいは無形成といった臨床症候を共有している。橈骨の低形成あるいは無形成は、RAPADILINO症候群とBGSではほぼ例外なく認められるが、RTSではそれらより出現頻度が低い。一方、多形皮膚萎縮はRTSの特徴であり、BGSでも報告例があるものの、RAPADILINO症候群ではみられない。ただし、多形皮膚萎縮は遅発性のため、1歳未満の段階では多形皮膚萎縮なしと断定することまではできない。頭蓋縫合早期癒合、特に冠状縫合の早期癒合は、BGSにとっては診断に係わる重要症候であるものの、RTSでは稀にしかみられない。脱毛症や睫毛・眉毛の欠損は、RTSの特徴的症候であるが、BGSやRAPADILINO症候群でこれがみられたとする例は今のところない。
鑑別診断
Baller-Gerold症候群(BGS)との鑑別を要する疾患として重要なものは、同一アレル疾患であるRothmund-Thomson症候群とRAPADILINO症候群(OMIM266280)である(「遺伝子の上で関連のある疾患」の項を参照)。
図1を参照されたい。
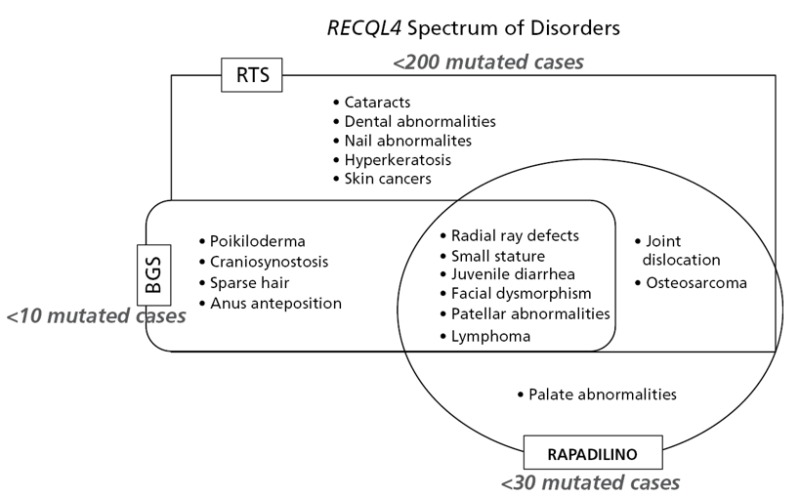
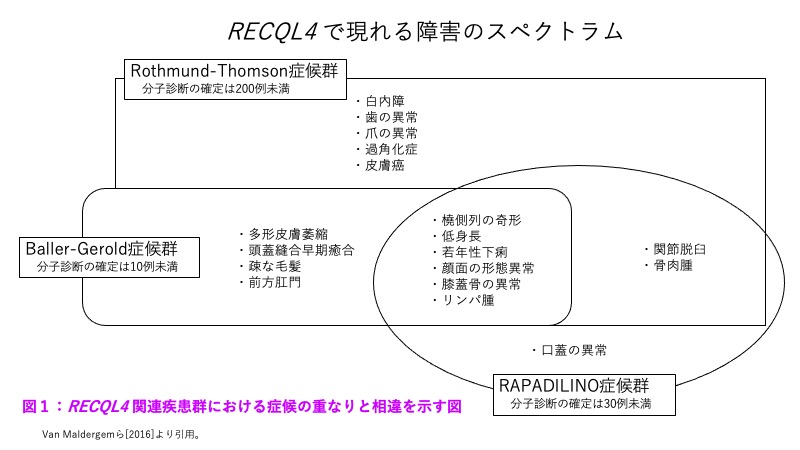
図1:RECQL4関連疾患群における症候の重なりと相違を示す図
注:「mutatedcases」とあるのは、分子診断がなされた例の意である。VanMaldergemら[2016]より引用。
鑑別を要するその他の疾患を表2に示した。
表2:BGSとの鑑別を検討すべき疾患
| 鑑別対象疾患 | 遺伝子 | 遺伝形式 | 鑑別対象疾患でみられる症候 | |
|---|---|---|---|---|
| BGSと重なる症候 | BGSと異なる症候 | |||
| Fanconi貧血(FA) | 多数1 | AR AD XL |
|
|
| 胎児バルプロ酸症候群(OMIM609442) | 関与なし | 関与なし |
|
|
| VACTERL(OMIM192350) | 不明 | 散発性 | 拇指低形成あるいは無形成 |
|
| SALL4関連疾患 | SALL4 | AD | 橈側列奇形 |
|
| Holt-Oram症候群 | TBX5 | AD | 橈骨に生じることのある上肢の奇形 |
|
| 血小板減少-橈骨欠損(TAR)症候群 | 脚注2参照 | 脚注2参照 | 時に重度であることもある上肢の短縮 |
|
| Saethre-Chotzen症候群 | TWIST1 | AD |
|
|
| Roberts症候群 | ESCO2 | AR |
|
|
| CDAGS症候群(OMIM603116) | 不明 | AR |
|
|
AD=常染色体顕性;AR=常染色体潜性;XL=X連鎖性
CDAGS=頭蓋縫合早期癒合と鎖骨低形成(craniosynostosisandclavicularhypoplasia);大泉門閉鎖遅延(delayedfontanelleclosure)と頭蓋骨欠損(cranialdefects)と難聴(deafness);肛門奇形(analanomalies);尿性器奇形(genitourinarymalformations);皮疹(skineruption)[Mendoza-Londonoら2005]
- FAの診断は、ジエポキシブタン(DEB)やマイトマイシンC(MMC)のようなDNA架橋剤を添加して培養を行った後の細胞で、染色体異常(切断,再構成,ラジアル構造化,転座)がみられることをもとに行われる。FA関連遺伝子として、約20の遺伝子が同定されている。
- 以前は常染色体潜性遺伝と考えられていたが、TAR症候群の遺伝形式は複雑で、この表現型が出現する上では、1q21.1の微小欠失が必要条件であるが、十分条件ではない[Klopockiら2007]。
臨床的マネジメント
最初の診断に続いて行う評価
Baller-Gerold症候群と診断された罹患者については、疾患の範囲を把握するため、すでに実施済でなければ、以下の事項が推奨される。
- 臨床遺伝医ないし遺伝カウンセラーとの面談
- 頭蓋縫合早期癒合の評価を目的とした脳神経外科医あるいは頭蓋顔面専門医による診察
- 手や腕の機能、ならびに手術の必要性をみるための整形外科的、作業療法的評価
- 多形皮膚萎縮が出現しているような場合は皮膚科的評価
症候に対する治療
頭蓋縫合早期癒合症の管理は、脳神経外科医/頭蓋顔面専門医の手で行う必要がある。頭蓋縫合早期癒合が両側性の場合は、通常、生後6ヵ月未満で手術が行われる。
拇指欠損例については、物をつかむ機能を回復することを目的として示指に対して行う拇指化手術で、満足すべき結果が得られたとの報告がみられる[Foucherら2005]。ただ、拇指無形成の子どもの多くは、整形外科的手術による介入なしでも機能は確保されている。
多形皮膚萎縮がみられる場合は、日焼け止めを上手に使うことで、紫外線への曝露に伴う皮膚癌のリスクから身を守ることができるように思われる。
がんが発生した場合は、当該のがんに詳しい腫瘍専門医のもとで治療を受ける必要がある。
定期的追跡評価
今のところ、BGS罹患者については、リンパ腫を発症した報告が1例存在するのみではあるが、RECQL4にRothmund-Thomson症候群ならびにRAPADILINO症候群関連の病的バリアントを有する例では、骨肉腫とリンパ腫に関し高リスクであることが知られている。そのため、潜在的リスクを考慮し、RECQL4の病的バリアントを有するBGS罹患者(あるいはその保護者)においては、こうした悪性腫瘍関連の徴候や症候にいつも気をつけておくことが望まれる。こうした徴候や症候の具体例としては、骨肉腫については骨痛、腫脹、跛行、リンパ腫についてはリンパ節腫脹や、発熱や原因不明の体重減少などの全身症候がある。
避けるべき薬剤/環境
理論上、皮膚癌のリスクが高まることが考えられることから、日光への過度の曝露は避ける必要がある。
リスクを有する血縁者の評価
リスクを有する血縁者に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「ClinicalTrials.gov」、ならびにヨーロッパの「EUClinicalTrialsRegister」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
Baller-Gerold症候群(BGS)は、常染色体潜性の遺伝形式をとる。
家族構成員のリスク
発端者の両親
- 罹患児の両親は、ともに絶対ヘテロ接合者(すなわち、病的バリアントを1つ有する保因者)である。
- ヘテロ接合者(保因者)は無症状で、本疾患を発症するリスクは有しない。
発端者の同胞
- 罹患者の同胞は、受胎の段階で、罹患者である可能性が25%、無症状の保因者である可能性が50%、罹患者でも保因者でもない可能性が25%である。
- ヘテロ接合者(保因者)は無症状で、本疾患を発症するリスクは有しない。
発端者の子
BGS罹患者の子は、病的バリアントを1つ有する絶対ヘテロ接合者(絶対保因者)となる。
他の家族構成員
発端者の両親の同胞は、保因者であることに関し50%のリスクを有する。
保因者の検出
リスクを有する血縁者に対して保因者の検査を行う上では、家系内に存在するRECQL4の複数の病的バリアントを事前に同定しておく必要がある。
関連する遺伝カウンセリング上の諸事項
家族計画
- 遺伝的リスクの確定、保因者であることの明確化、出生前/着床前遺伝学的検査を受けるかどうかの話し合いに最も適しているのは、妊娠前の時期である。
- 罹患者、保因者である若い成人、ならびに保因者であることに関しリスクを有する若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法であるとか、遺伝子・病原のメカニズム・疾患等に対するわれわれの理解が、将来はより進歩していくことが予想される。そのため、分子診断の確定していない(すなわち、背景にある病原のメカニズムが未解明の)発端者については、DNAの保存を検討すべきである。詳細な情報については、Huangら[2022]を参照されたい。
出生前検査ならびに着床前遺伝学的検査
分子遺伝学的検査
家系内に存在するRECQL4の病的バリアントが特定されている場合は、出生前検査や着床前遺伝学的検査を行うことが可能である。
超音波検査
連続的に行う超音波検査で、四肢の短縮、橈骨の低形成/無形成、頭の形の異常(短頭症)が明らかになることがある。BGSのリスクを有する妊娠において、妊娠14週以降に行った超音波検査で上記のような所見がみられたことで、BGSの存在が明らかになったとする報告がみられる[VanMaldergemら1992,Siitonenら2009,Caoら2015]。
出生前検査の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。
現在、多くの医療機関では、出生前検査を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- Children'sCraniofacialAssociation
- FACES:NationalCraniofacialAssociation
- NationalInstituteofNeurologicalDisordersandStroke(NINDS)
- REACH
Phone:800-535-3643
Email:contactCCA@ccakids.com
www.ccakids.org
Phone:800-332-2373;423-266-1632
Email:info@faces-cranio.org
www.faces-cranio.org
Phone:800-352-9424
Craniosynostosis
Helpingchildrenwithupperlimbdifferenceslivelifewithoutlimits.
UnitedKingdom
Phone:08451306225;02034780100
www.reach.org.uk
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:Baller-Gerold症候群:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specificデータベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| RECQL4 | 8q24.3 | ATP依存性DNAヘリカーゼQ4 | RECQL4 database | RECQL4 | RECQL4 |
データは、以下の標準資料から作成したものである。
遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:Baller-Gerold症候群関連のOMIMエントリー(内容の閲覧はOMIMへ)
| 218600 | BALLER-GEROLD SYNDROME; BGS |
| 603780 | RECQ PROTEIN-LIKE 4; RECQL4 |
分子レベルの病原
BGSは、RECQL4の病的バリアントによって生じる疾患である。RECQL4は、RecQの名で知られるヘリカーゼスーパーファミリーⅡに属し、1,208のアミノ酸から成るタンパク質、ATP依存性DNAヘリカーゼQ4をコードしている。RecQは、2本鎖のDNAやRNA-DNAハイブリッドを解いて、1本鎖DNAの鋳型を産生するときの3’-5’極性に従って分類されている。RECQL、BLM、WRN、RECQL4、RECQL5という少なくとも5つのRecQヒトパラログが知られており、それぞれ独自の役割を担いつつも、一部の役割は重なる。BLMの病的バリアントはBloom症候群を、WRNの病的バリアントはWerner症候群を引き起こす。両疾患とも、常染色体潜性遺伝の染色体不安定疾患で、がんの素因ないし早老症を特徴とする。これらの症候は、RECQL4関連常染色体潜性RTSや、遺伝的不安定性、発育不全、がんの素因などの表現型上の特徴が互いに重なるRAPADILINO症候群とBGSでもみられる。RECQL、RECQL5については、現在のところ、これに関連してヒトに生じる疾患の報告はみられない。
RecQヘリカーゼは、DNAのプロセシングに関するさまざまな段階(複製,組換え,修復,テロメアの維持)においてだけでなく、翻訳、RNAプロセシング、mtDNAの維持、染色体の分離といったことにも重要な機能を有している[Croteauら2014]。RecQヘリカーゼは、実質的に、DNA代謝の全局面で作用していることから、その発現や生化学的活性の乱れはゲノムの不安定性となって現れ、疾患やがんの素因を生じる結果となる[Bochman2014]。
遺伝子構造
RECQL4は21のエクソンをもち、そのサイズは6.5kbを超える。この遺伝子には3,627bpのコーディング配列があり、その発現は、転写因子Sp1とAP2との結合部位を含むハウスキーピングプロモーターによる調節を受けている。これがコードするのは、1,208のアミノ酸から成る133kdのタンパク質、RECQprotein-like4(RECQL4)で、このタンパク質には、大腸菌RecQヘリカーゼと相同のDNAヘリカーゼドメインのエクソンをもつが、これはヒトのRecQファミリーに属する5つのメンバーすべてが共通して有している。RECQL4のヘリカーゼドメインは、ATP結合ドメイン(アミノ酸残基489-662)とC末端ドメイン(アミノ酸残基683-850)も含め、エクソン8-15によりコードされている。RECQL4は100bp未満のイントロンを13個有することが特徴的で、これによりスプライシングの進行が遅くなりやすくなる[Wangら2002]。遺伝子とタンパク質に関する情報の詳細は、表Aの「遺伝子」の欄を参照のこと。
病的バリアント
これまでに、7家系で11種の病的バリアントが同定されている。
- c.3056-2A>C(ホモ接合)[Mégarbanéら2000,VanMaldergemら2006]
- c.3061C>T(p.Arg1021Trp)とc.1573del―この複合ヘテロ接合を2家系で確認[VanMaldergemら1992,VanMaldergemら2006,Siitonenら2009]
- c.2335_2356del(ホモ接合)[Siitonenら2009]
- c.496C>T(p.Gln166Ter)とc.3151A>G(p.Ile1051Val)―この複合ヘテロ接合を妊娠中絶の2例で確認[Siitonenら2009]
- c.2492_2493delとc.2506_2518del―この複合ヘテロ接合を、重度のBGS表現型をもち2.5歳時に正中NK/Tリンパ腫を発症した1小児例で確認[Debeljakら2009]
- c.2059-1G>Cとc.2141_2142del―この複合ヘテロ接合を重症BGS(出生前診断)の1胎児で確認[Caoら2015]
- イントロン12からエクソン18までの欠失(1,614bp欠失と1bpのGの挿入;g.145737562_145739175delinsC)―このホモ接合を、カフェオレ様の斑を有するものの多形皮膚萎縮は示さない4歳の日本人男児で確認[Kanekoら2017];ⅣからⅥまでのヘリカーゼモチーフ欠失が予測される
2つあるp.Arg1021Trpとp.Ile1051Valのミスセンスバリアントが、ともに、RecQのC末端領域(RQC)部に生じたものであることは、興味深いところである。この領域は、RECQL4だけでなく、そのパラログにとっても、適切な機能を発揮する上で不可欠の部分である[Mojumdarら2017]。
現在までに報告されているRECQL4の11種の病的バリアントのうち、
- 7つはBGSのみで検出されている。
- p.Arg1021Trpとc.2492_2493delの2つは、BGSとRTSでみられる。
- 反復性のc.1573delは、BGS、RTS、RAPADILINOで確認されている。
- c.2059-1G>Cのバリアントは、BGSの胎児に加え、RTSの1例でも報告されている[Beghiniら2003]。
これと同じ部位の別の塩基への変化が、RTS(c.2059-1G>T)[Kitaoら1999,Kellermayerら2005,Siitonenら2009,Piardら2015]でも、またRAPADILINO(c.2059-1G>A)[Siitonenetal2009]でも報告されている点は注目に値する。
分子診断での確認が行われた非血縁例が少数であるにもかかわらず、複合ヘテロ接合が半数以上(ホモ接合が3例であるのに対し複合ヘテロ接合が4例)に認められている。
これは、罹患者の約3分の2が複合ヘテロ接合である[VanMaldergemら2016]というRTSでみられる傾向に一致するものである。
表3:本GeneReviewで取り上げたRECQL4の病的バリアント
| DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | 参照配列 |
|---|---|---|
| c.496C>T | p.Gln166Ter | NM_004260.3 NP_004251.3 |
| c.1573del | p.Cys525ALAfsTer33 | |
| c.2059-1G>C | p.? | |
| c.2059-1G>T | p.? | |
| c.2059-1G>A | p.? | |
| c.2141_2142del | p.Glu714AlafsTer94 | |
| c.2335_2356del | p.Asp779CysfsTer57 | |
| c.2492_2493del | p.His831ArgfsTer52 | |
| c.2506_2518del | p.Ser836TrpfsTer3 | |
| c.3056-2A>C | p.? | |
| c.3061C>T | p.Arg1021Trp | |
| c.3151A>G | p.Ile1051Val | |
| g.14573562_145739175delinsC | NC_000008.10 |
上記のバリアントは報告者の記載をそのまま載せたもので、GeneReviewsのスタッフが独自に変異の分類を検証したものではない。GeneReviewsは、HumanGenomeVariationSociety(varnomen.hgvs.org)の標準命名規則に準拠している。
命名規則の説明については、QuickReferenceを参照のこと。
正常遺伝子産物
RECQL4によってコードされるタンパク質、ATP依存性DNAヘリカーゼQ4は、その主要な機能として、DNAの複製や修復に際して生じる異常な構造をもつDNAの処理を担っていると考えられている。変異に伴って安定な二次構造ができ上がることで、DNA複製フォークの移動が障害を受けることは、複製フォークの進行阻害につながり、結果としてフォークの停止や崩壊、さらには異常構造物の排除障害を経て、染色体の不安定性へと、そして最終的には細胞死あるいはがんへとつながっていく。こうした意味で、ATP依存性DNAヘリカーゼQ4は、ゲノムの番人であるとも言えよう[Wu&Hickson2006]。
RECQL4は非常に多機能なタンパク質で、その生物学的役割については、他のRecQファミリーと一部が相互に関連し合う関係にある。このタンパク質のもつヘリカーゼ活性は、かつて疑問視された時期もあったものの、現在では完全に証明されており、RECQL4の保存されたヘリカーゼモチーフと、アミノ酸240から400までのN末端ドメインが、それぞれ独立した形でATP依存性DNA巻き戻しを促進することでこの機能を担っていることがわかっている[Xu&Liu2009]。また、RecQのC末端領域(RQC)は、他のすべてのヒトパラログにおいて触媒コアを構成する不可欠の一部分であるが、この部分が物理的に欠損するという不可解な謎は、現在では、ヒトRECQL4内に機能的RQCドメインが存在することが実験によって実証されたことで解決している[Mojumdarら2017]。すべてのヒトRecQ群と同様、RECQL4もDNAの組換えや修復に不可欠であり[Croteauら2014]、BLMやWRNと協調し合ってテロメア維持の役割を果たしている[Ghoshら2012]。DNAの複製に関して、RECQLとRECQL4は、一緒に複製複合体を構成する要素であり、DNAの複製開始や複製フォークの移動に関し、それぞれ別々の役割を果たしている[Xu&Liu2009,Thangavelら2010]。RECQL4のヘリカーゼ活性は、進行の止まった複製フォークに対するレスキューに特化した働き、あるいは複製開始点における最初の巻き戻しの働きをしているのではないかとの考えが提唱されている[Masai2011]。
RECQL4に絶対的に特異的な働きとして、ミトコンドリアDNAのコピー数の維持、ならびにp53(この遺伝子の病的バリアントによりLi-Fraumeni症候群が生じる)のミトコンドリアへの輸送に関する調節が挙げられる[Deら2012]。RECQL4とp53の両方が働くことでポリメラーゼγの活性が生まれ、ヒトミトコンドリアゲノムの健全性が維持される[Guptaら2014]。最後になるが、RECQL4の欠陥に関連して生じるがん素因に関して何より重要なこととして、N末端則経路に関与するユビキチンリガーゼであるUBR1やUBR2[Yinら2004]との安定した相互作用を通じて、染色体分離の正しい進行にRECQL4が決定的に重要な役割を果たしていることが挙げられる[Raoら2001]。RECQL4とコヒーシン経路との間に相互接続の関係があることは、CorneliadeLange症候群罹患者の細胞内でRECQL4の下向き調節が生じていること[Liuら2009]、ならびに、コヒーシン経路内で作用する付属タンパク質群としてRECQL4が含まれるといった事実により証明されている。
異常遺伝子産物
同定済のRECQL4の病的バリアントの大多数はフレームシフトバリアントあるいはナンセンスバリアントで、成熟mRNAの不安定化、トランケーション型タンパク質の低レベルでの産生といったことを予測させるものとなっている[Ouyangら2008]。N末端が維持されることはDNA複製の開始に決定的に重要であり、細胞の生存にとって欠くことのできないものであるように思われる。すべてのRECQL4関連疾患を通じて、この領域には病的バリアントがほとんどみられない(ある場合でもホモ接合ではない)という事実が、このことを裏書きしている。変異RECQL4タンパク質の機能研究は、まだ限られた数しか行われていないものの、RECQL4の細胞における機能がヘリカーゼ依存性であることを支持するものとなっており[Croteauら2012]、同時に、ヘリカーゼ活性を損なうバリアントと骨肉腫との間に関連性があるのではないかとする考え方を確認するものとなっている[Wangら2003]。
更新履歴:
-
Gene Reviews著者: Lionel Van Maldergem, MD, PhD, Juliette Piard, MD, Lidia Larizza, MD, and Lisa L Wang, MD.
日本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2018.4.19. 日本語訳最終更新日: 2023.5.28.[in present]
![]()