





大脳白質障害の臨床診断について
国立精神・神経医療研究センター病院小児神経科
佐々木征行
1.概念・定義
一次性大脳白質障害は、主に中枢神経の髄鞘を障害する疾患である。遺伝性疾患が多く、変性疾患・代謝性疾患などからなる。大脳白質変性症、脱髄性疾患、髄鞘低形成などが含まれる。
2.病因
原因遺伝子が確定している疾患もあれば未確定の疾患もある。
古典的な分類として、中枢神経の髄鞘を形成する蛋白質(プロテオリピッド蛋白:PLP, ミエリン塩基性蛋白:MBP)、細胞内小器官(ペルオキシソーム、ライソゾーム、ミトコンドリアなど)、代謝関連物質(アミノ酸代謝異常、有機酸代謝異常)などで分類し、その他として細胞内での様々な機能蛋白質をコードする遺伝子の異常が加えられている。(表1)
表1.一次性大脳白質障害の古典的分類と代表的疾患
- 髄鞘蛋白異常症
Pelizaeus-Merzbacher病 (Proteolipid protein; PLP)
18q- syndrome (Myelin basic protein; MBP) - Lysosome病
Krabbe病(Globoid cell leukodystrophy; GLD)
異染性白質ジストロフィー (MLD)
GM1/GM2 gangliosidosis - Peroxysome病
副腎白質ジストロフィー (ALD)
Zellweger syndrome - Mitochondria病
Kearns-Sayre syndrome
Progressive cavitating leukoencephalopathy - アミノ酸代謝異常症
フェニルケトン尿症
尿素サイクル異常症 - 有機酸代謝異常症
Canavan病
L-2-hydroxyglutaric aciduria - その他の代謝異常症、変性疾患
Alexander病
Hypomyelination with atrophy of basal ganglia and cerebellum (H-ABC)
Pol III-related leukodystrophies (4H syndrome, ADDH, TACH, LO, HCAHC)
Megalencephalic leukoencephalopathy with subcortical cysts (MLC)
Leukoencephalopathy with vanishing white matter (VWM)
Hypomyelination with congenital cataracts (HCC)
3.臨床症状
疾患によって発症時期は異なる。症状は、運動発達遅滞あるいは運動退行、痙性麻痺、筋緊張亢進、知的障害(遅滞、退行)などが基本である。これに、けいれん発作、小脳性失調、不随意運動(ジストニアなど)、内分泌障害(低身長、性成熟の遅れ)などを合併することがある。
4.病態
(1)病理学的分類
- 脱髄(demyelination)は、一旦完成した髄鞘が喪失することである。脱髄は中枢神経系にも末梢神経系にも起き得る。
- 髄鞘低形成(hypomyelination)は、髄鞘形成成分が不完全であるため機能的に不完全な髄鞘が存在し、早期に崩壊しやすくなる。髄鞘異形成(dysmyelination)もほぼ同義として使用される。
(2)MRIによる分類
病変部の大脳白質の信号強度を、T1強調画像とT2強調画像で比較したり、大脳皮質の信号強度と比較したりすることによって、脱髄か髄鞘低形成かにある程度区別できる1, 2)。 1)脱髄(demyelination)では、病変部の白質はT2強調画像で強度高信号でありかつT1強調画像で強度低信号を呈する。一般的に病初期には皮質下白質(U-fiber)は保たれる。 2)髄鞘低形成(hypomyelination)では、病変部の白質は大脳皮質と比較してT2強調画像で軽度高信号であり、かつT1強調画像で高信号(正常パターン)・等信号・軽度低信号と様々な信号強度を呈す。皮質下白質から脳室周囲の深部白質まで同程度の信号強度であることが多い。
この基本画像に加えて、大脳白質が量的に増加・減少したり、透明化・嚢胞化したり、造影されたり、石灰化を認めたりすることもあり、鑑別診断に有用となる。
5.診断と鑑別診断
発症時期と症状と頭部MRI画像所見からある程度の鑑別が可能である。
大脳白質のどの部位から(前頭葉優位か頭頂葉優位かなど)信号異常が始まるかが診断に役立つことがある。
A.脱髄性疾患
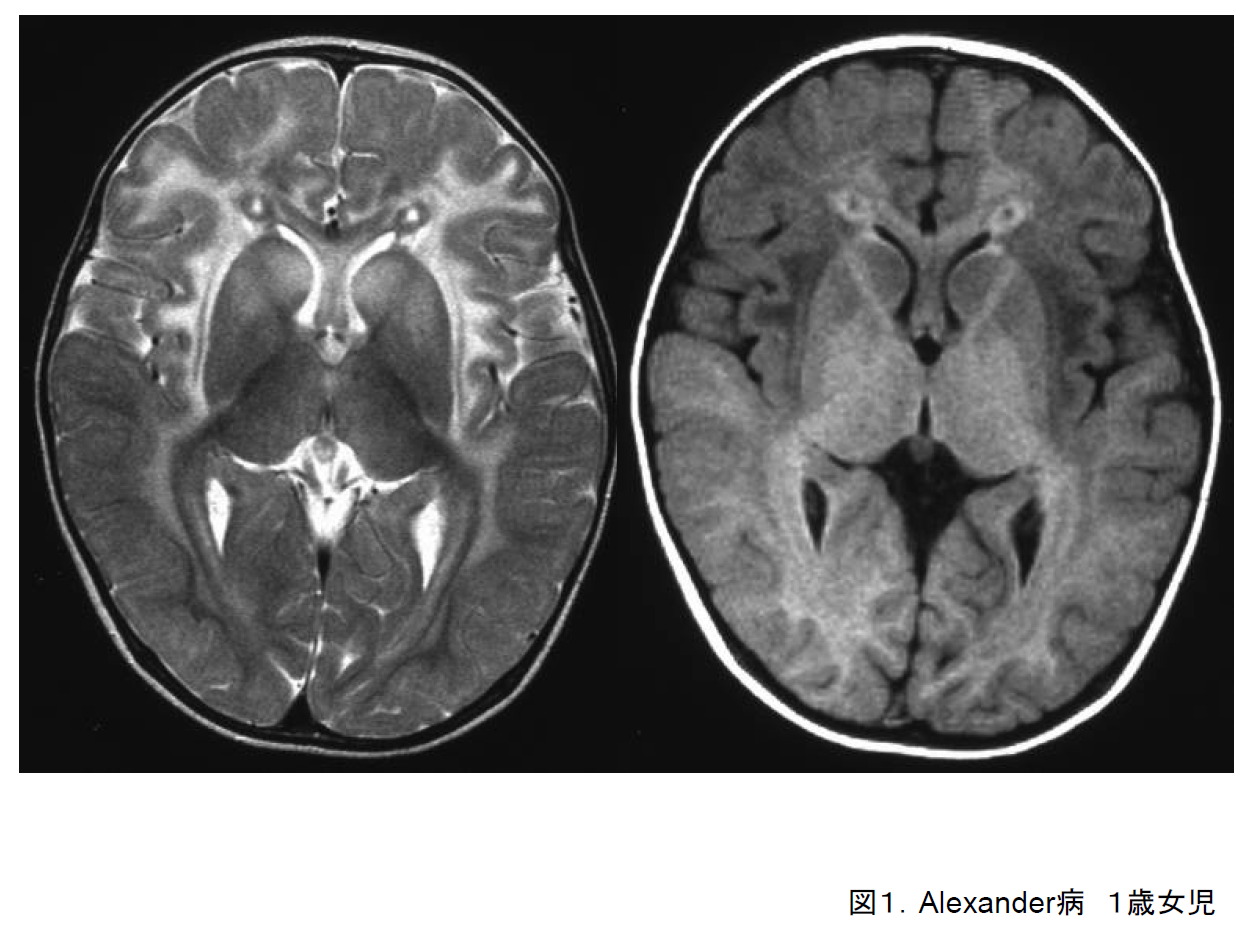
(1)Alexander病
大脳優位型では、頭囲拡大、けいれん、精神運動発達遅滞が主症状である。1歳前には発症することが多い。進行すると痙性四肢麻痺、嚥下障害を呈する。
頭部画像では前頭葉白質から異常部位が拡がる(図1)。異常信号部位では白質量が増大しているように見えることが多い。
脳幹優位型は成人に多く大脳白質の信号異常は一定しない。
GFAP遺伝子の変異で発症し常染色体優性遺伝形式を示す。大脳優位型では、基本的には新生突然変異である。遺伝子診断ができるようになってからは、脳生検によってRosenthal線維を確認する必要はなくなった。
(2)副腎白質ジストロフィー(X-linked adrenoleukodystrophy; ALD)
小児大脳型は3歳から10歳の間に視力異常、行動異常、学業成績低下、歩行障害、けいれんなどで発症する。数年で四肢麻痺により常時臥床まで進行することが多い。
発症時期によって思春期大脳型、成人大脳型の分類がある。
大脳型、脊髄型などがあり、同じ変異を持つ同一家系内でも発症時期や病型が異なることがある。頭部MRIでは側脳室後角周囲の後頭葉・頭頂葉深部白質から脱髄が始まることが多い(図2-1)。ときに前頭葉から始まる場合もある(図2-2)。
Xq28に存在するABCD1遺伝子異常により発症し、性染色体劣性遺伝形式を示す。
血中極長鎖脂肪酸増加が診断上重要である。
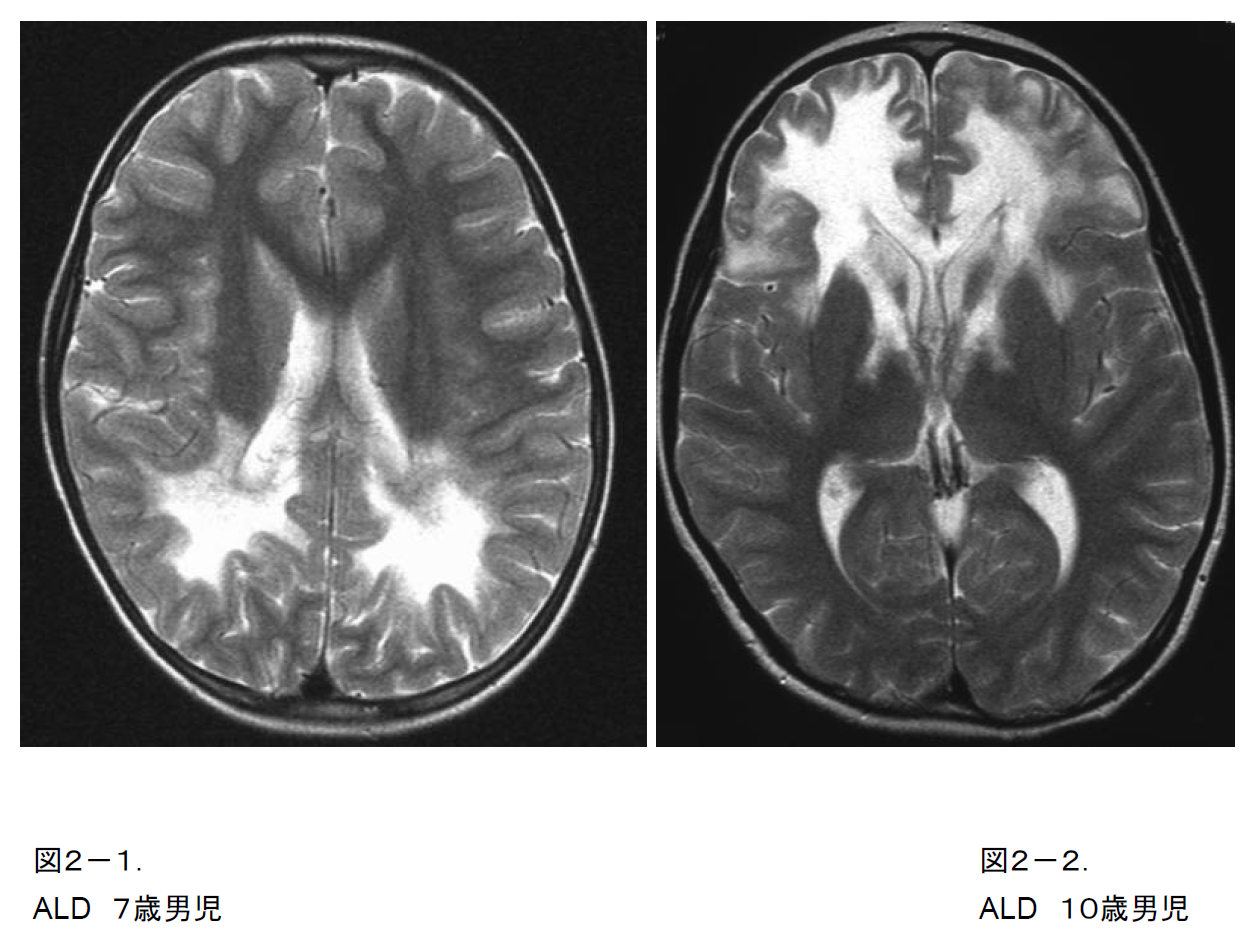
図2-2. ALD.10歳男児.T2強調画像.前頭葉と内包前脚から始まる非典型例.
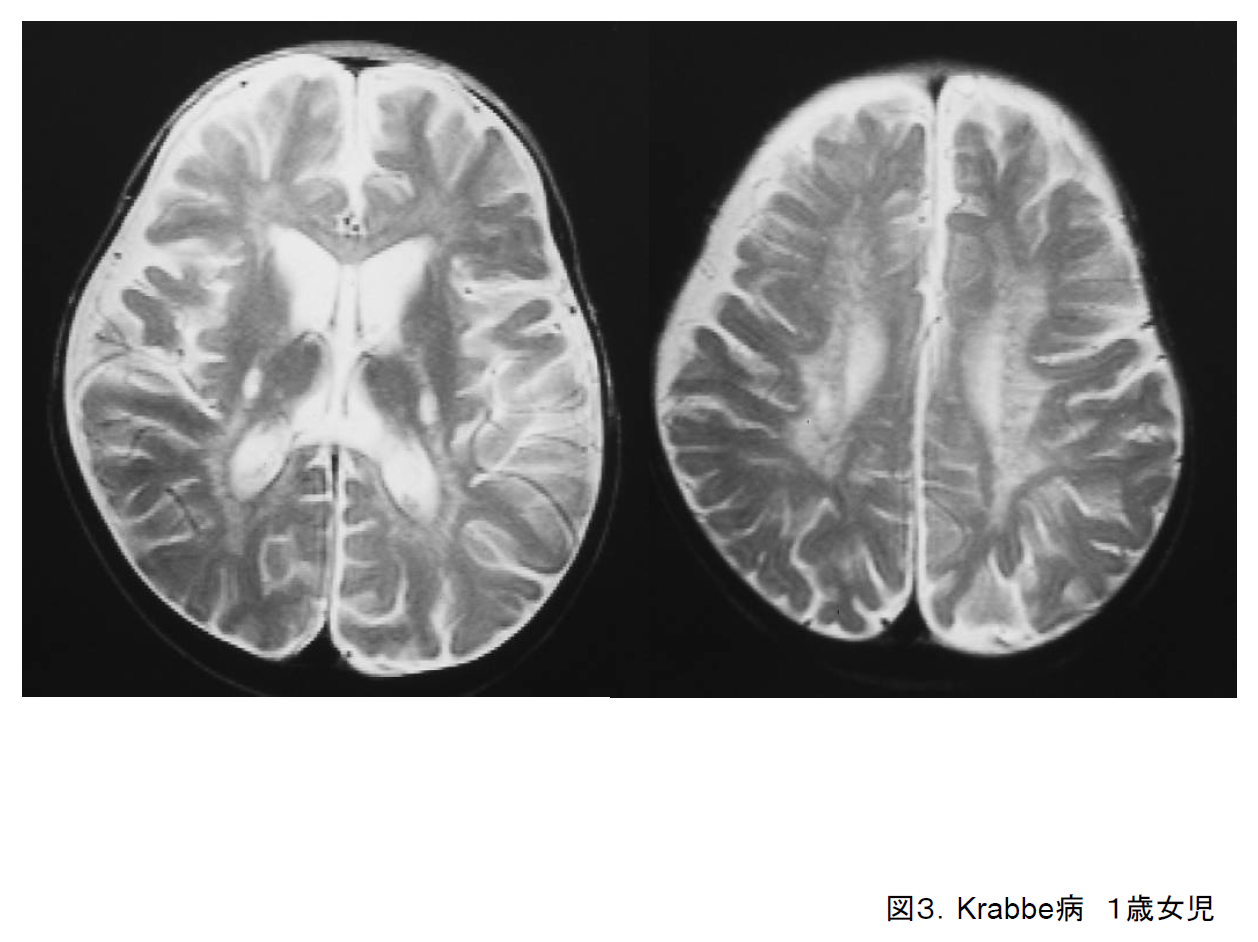
(3)Krabbe病(GLD)
乳児型は生後6か月以内に頚定不安定、哺乳不良、易刺激性亢進などで気付かれる。末梢神経障害を伴うために、深部腱反射の低下・消失が大きな特徴である。急激に進行し、1歳くらいまでには常時臥床となる。乳児後期型から成人型まで発症時期は幅広い。
頭部MRIでは側脳室後角周囲から後頭頭頂葉白質に脱髄が拡がる。(図3)、内包後脚や小脳髄質にも異常を認めることが多い3)。
病歴、画像、末梢神経伝導検査の異常から疑う。最終診断は、ガラクトセレブロシダーゼ活性低下とGALC遺伝子変異を確認する。
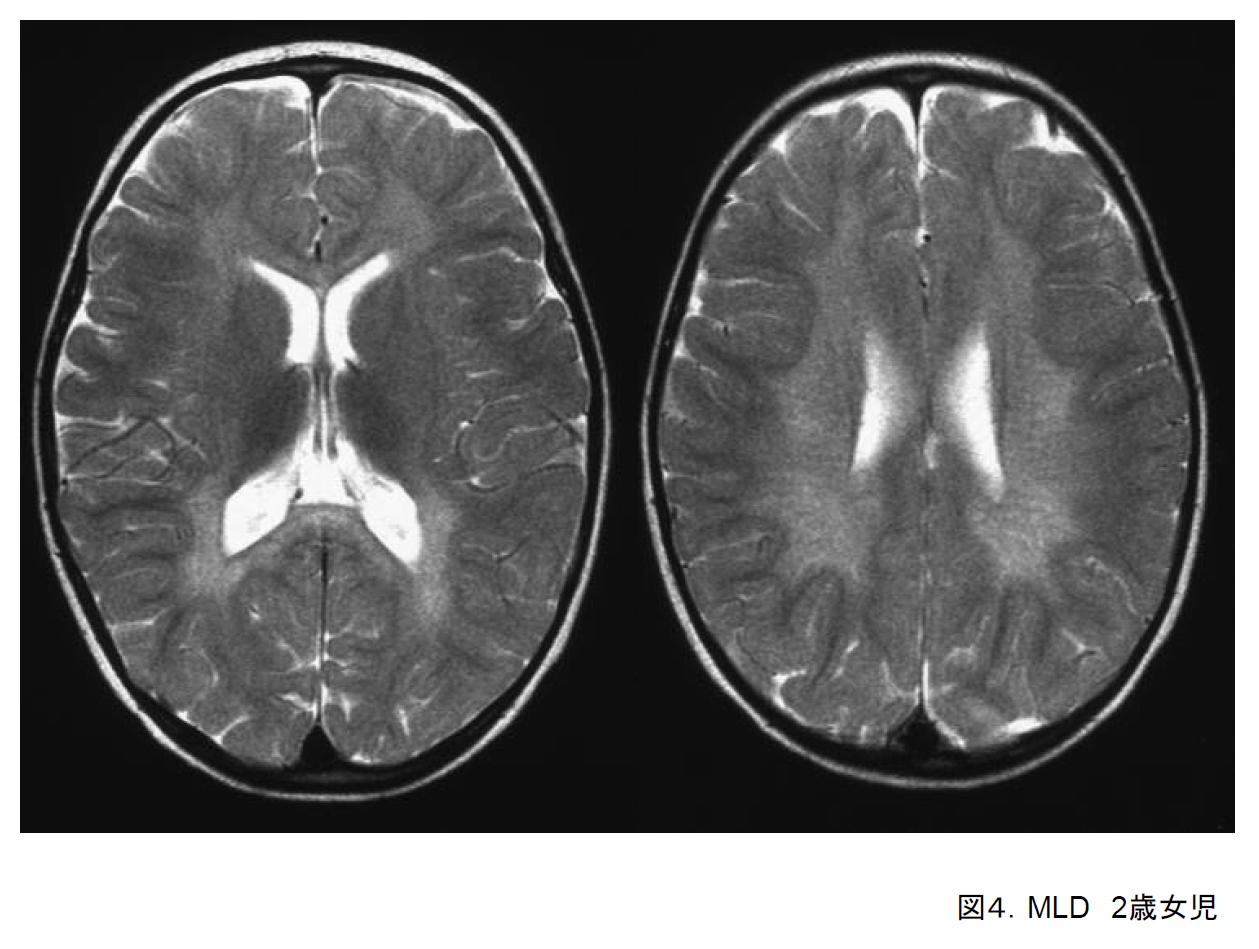
(4)異染性白質ジストロフィー(MLD)
乳幼児型では1歳半から2歳までの時期に、転びやすくなる、筋力低下、言葉が減る、などの症状で気付かれる。痙性麻痺、強直けいれん発作などを呈し、数年以内に常時臥床状態になる。本症でも末梢神経障害のために深部腱反射は低下・消失する。発症時期により若年型や成人型もある。
頭部MRIでは、前頭・頭頂・後頭葉の深部白質に脱髄が拡がる(図4)。
診断は、病歴、頭部画像所見と末梢神経伝導検査の異常で疑われたら、Aryl sulfatase Aの活性低下とARSA遺伝子変異を確認する。
(5)Megalencephalic leukoencephalopathy with subcortical cysts (MLC)
乳児期早期から大頭を認める。精神運動発達は軽度の遅れを示す。筋緊張低下や不安定歩行を示しやすい。けいれん発作を伴うこともある。
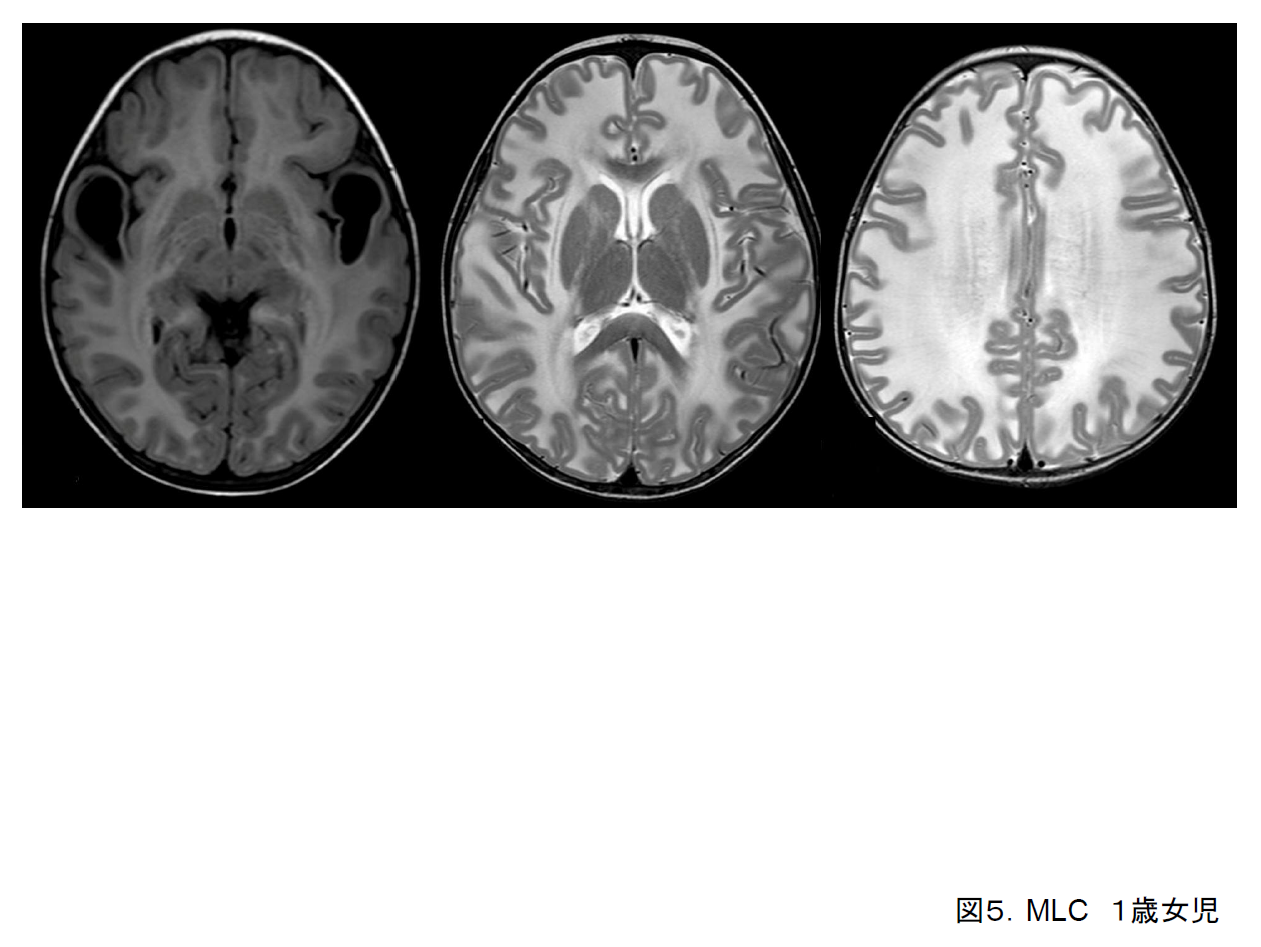
頭部MRI画像では全般的な大脳白質量の増加とT2強調画像でも高信号を基本とし、側頭葉前部や前頭・頭頂葉皮質下白質に嚢胞を形成しやすい(図5)。本疾患も頭部画像所見から独立疾患とされ責任遺伝子(MLC1)も同定された4)。
(6)Leukoencephalopathy with vanishing white matter (VWM)
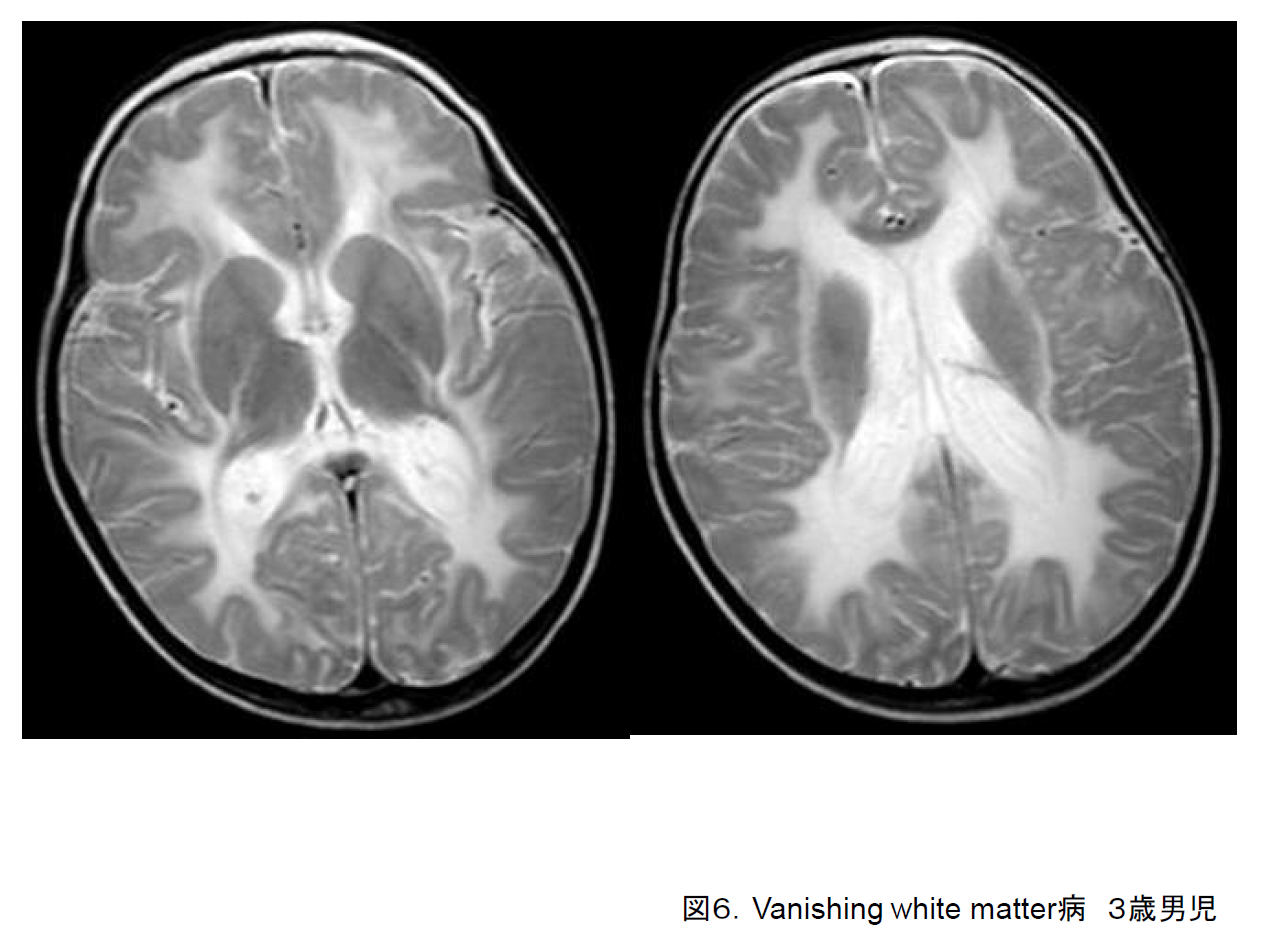
Vanishing white matter病ともいう。発症は1歳から成人まで広い。感染症や軽微な頭部外傷などで階段状に運動退行および知的退行が進行することが多い5)。T2強調画像で高信号を示している大脳白質(図6)が、次第にFLAIR画像で低信号となり脳脊髄液と同様に変化する。EIF2B1-5遺伝子の異常が報告されている。
B.髄鞘低形成
(1)Pelizaeus-Merzbacher病(PMD)
乳児期に、眼振(振り子様あるいは回転性)、精神運動発達遅滞、筋緊張低下などで気付かれる。小脳症状(失調、振戦など)、大脳基底核症状(固縮、ジストニアなど)あるいは強直けいれん発作などを合併することもある。ある程度遺伝子型と臨床型との相関が知られている。先天(Connatal)型では頚定も困難であり、乳幼児期に死亡することもある。古典(Classic)型では頚定はかなり遅れて獲得して比較的高齢まで生存する例(20~60歳)も多い。
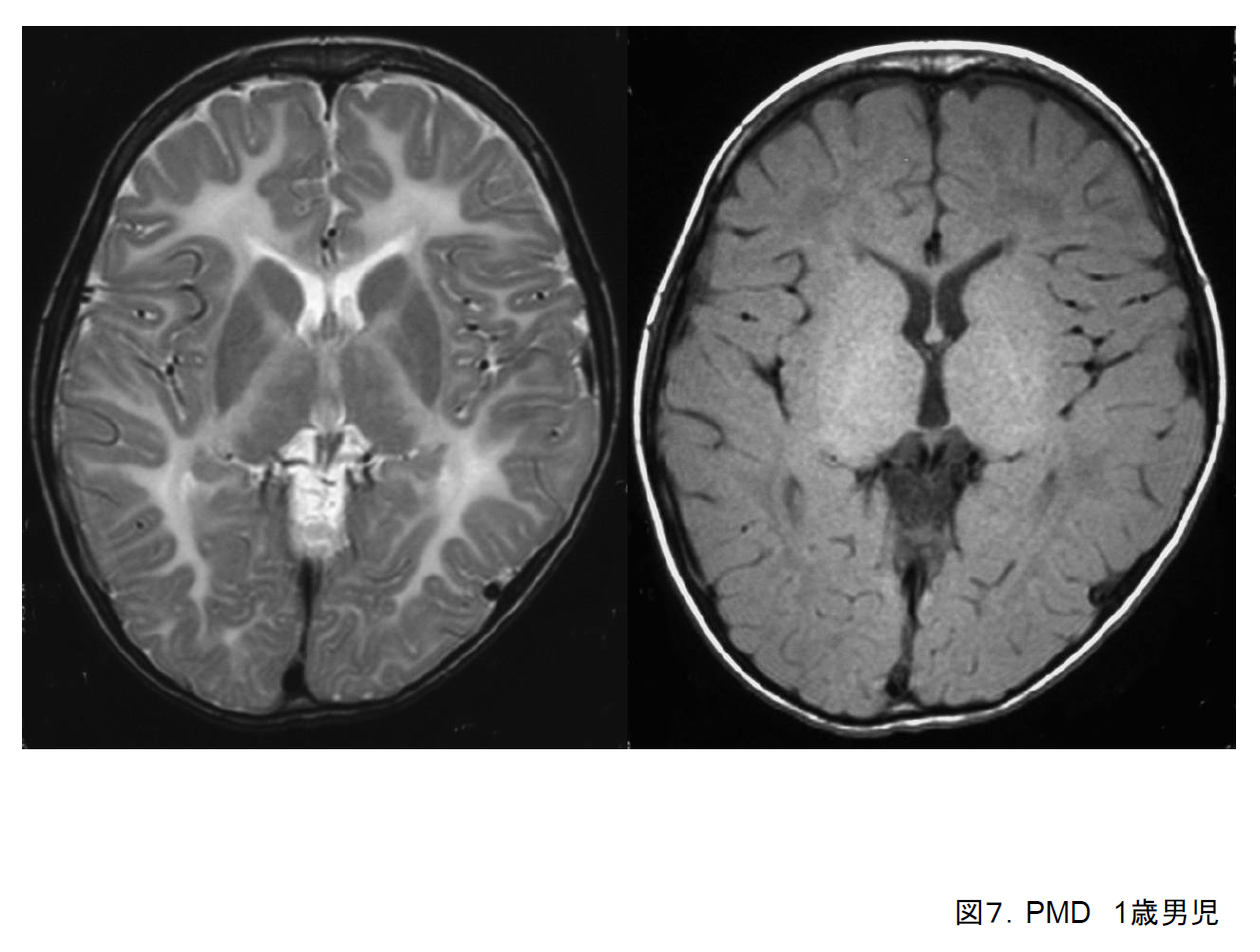
頭部MRIでは、先天型ではT2強調画像では髄鞘はほぼ全て高信号でほとんど形成されない(図7)が、T1強調画像では等信号のことが多い。中には軽度高信号を示して髄鞘が形成されているように見えることもある。
X染色体上のPLP1遺伝子の重複あるいは変異で発症し、性染色体劣性遺伝形式を示す。男児で、眼振、頭部画像での低髄鞘化、および聴性脳幹反応(ABR)でIII波以降の消失があれば強く疑う。
(2)Pelizaeus-Merzbacher様病(PMD-like disease)
乳児期より眼振と精神運動発達遅滞を呈し頭部MRI画像所見も含めて臨床的にPMDと診断されて、PLP1遺伝子に異常が出ない群がある。女児例も含まれる。常染色体劣性遺伝形式をとり、GJC2遺伝子異常が見出された。
(3)18q-症候群
乳幼児期に精神運動発達遅滞、筋緊張低下、低身長、などに気付かれる。顔貌異常(顔面正中部低形成、くぼんだ眼球、眼裂狭小、鯉様の口など)を示すこともある。特異的な症状はない。
頭部MRIではT2強調画像で大脳白質全体の高信号を認める例から散在性に高信号を認める例まで多彩であり、画像所見からの診断は難しい。
染色体G-bandで18番染色体長腕端部に欠失を認めることで診断される。髄鞘化の異常はミエリン塩基性蛋白質(MBP)遺伝子のハプロ不全によると想定されている。
(4)Hypomyelination with atrophy of basal ganglia and cerebellum (H-ABC)
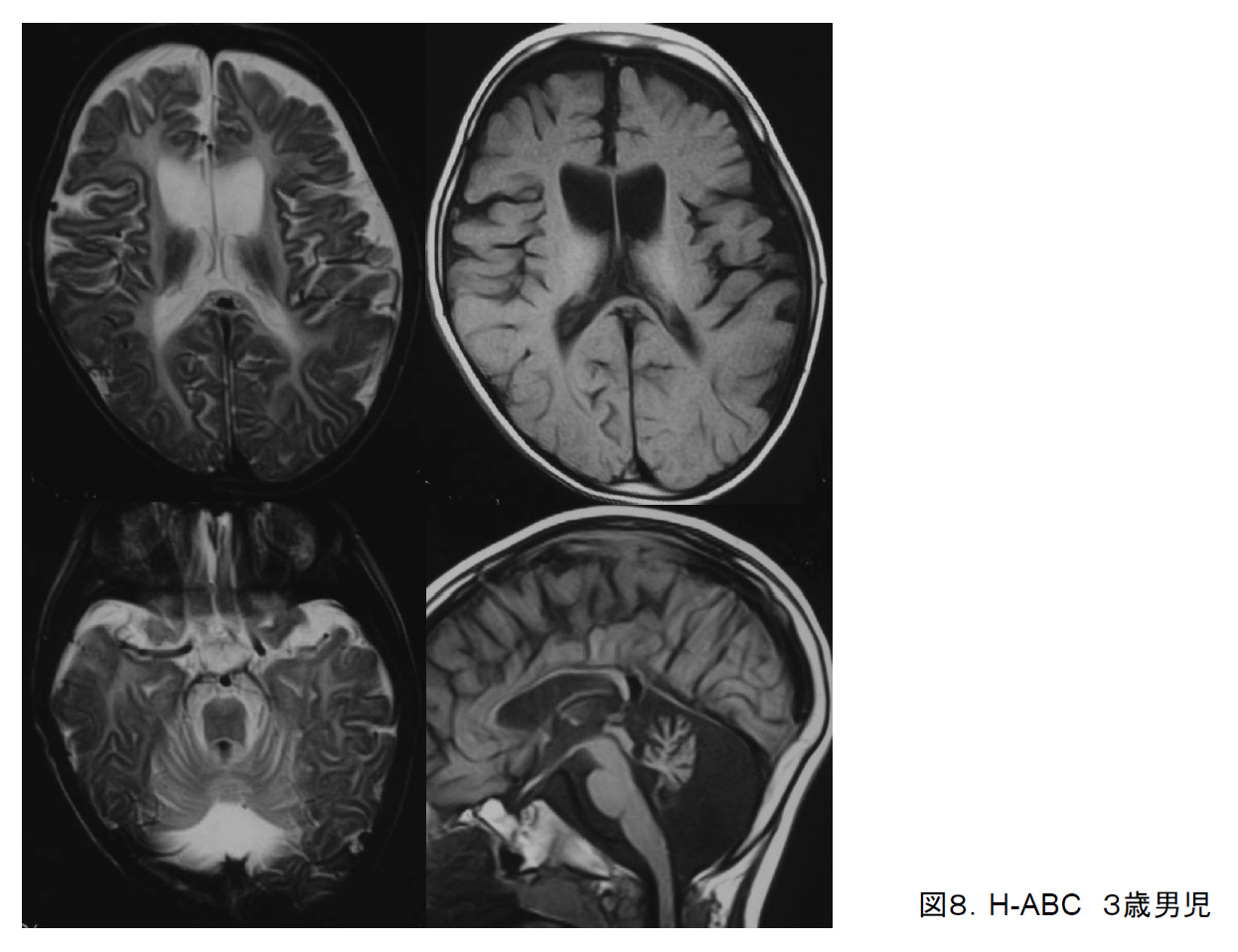
幼児期より歩行障害、ジストニア、小脳失調などを呈し、緩徐進行性である。早期からジストニアを呈することが特徴である。大脳白質低形成に加えて両側基底核と小脳の萎縮を示す画像所見(図8)から独立疾患として報告され、責任遺伝子(TUBB4A)が見出され、特定の変異(D249N)に限られることが分かった6)。基底核特に尾状核と被殻が著明に萎縮してT2強調画像で高信号を呈することが特徴的であり、これが髄鞘低形成とともに認められる場合はH-ABCの診断は難しくない。
同じTUBB4A遺伝子の中にH-ABCとは異なる変異をもち、小脳萎縮はあるものの被殻が強く萎縮せず信号変化も示さない病型(図9)7)や、不随意運動症だけを呈し頭部MRIで異常を示さない病型(DYT4)も見出されている。これらを含めてTUBB4A関連疾患と提唱されている。
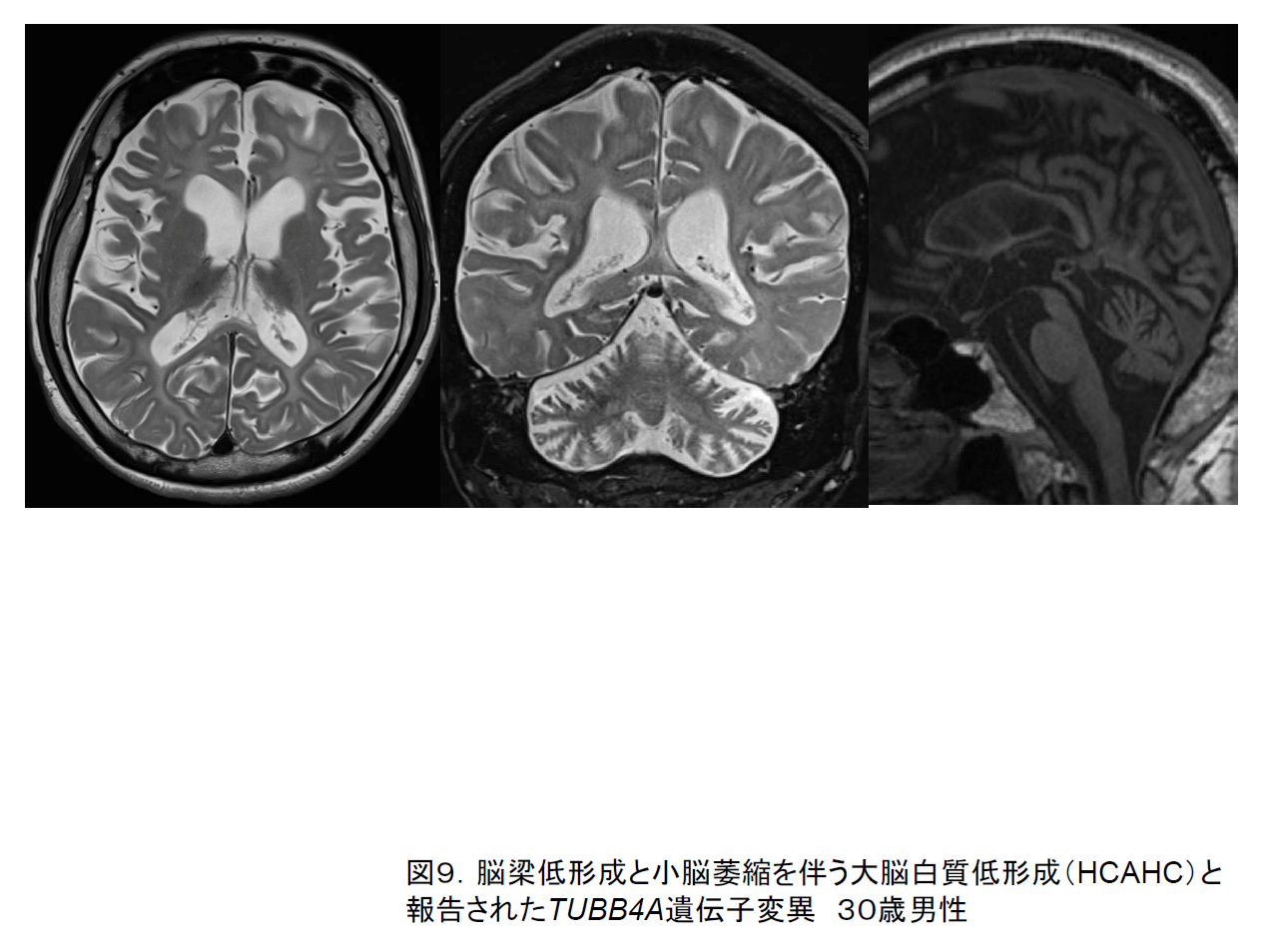
(5)Pol III関連白質ジストロフィー(Pol III-related leukodystrophies)
頭部MRIで全般的な大脳白質低形成を認めても特異的診断名をつけることが困難なことがしばしばある。これらの中で、小脳萎縮と脳梁低形成の組み合わせをもつ一群がある。これらは、痙性あるいは小脳失調による進行性歩行異常、振戦を基本症状として示し、他の症状つまり異常な歯牙(歯が少ない、萌出の遅れ)や下垂体性の性腺機能低下症(性成熟の遅延/欠損)などの組み合わせによって、4H (Hypomyelination, hypodontia, hypogonadotropic hypogonadism) syndrome8)、ADDH (Ataxia, delayed dentition, and hypomyelination)、TACH (Tremor-ataxia with central hypomyelination)、LO (Leukodystrophy with oligodontia)、HCAHC (Hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum) 9)などの診断名で報告されてきた。いずれも1,2歳で症状が顕在化するも進行は緩徐で通常不安定ながら歩行を獲得する。責任遺伝子(POLR3A/POLR3B)が同定され10, 11, 12)、これらが一連の表現型の違いによることが解明され、Pol III関連白質ジストロフィーと提唱されている13)。
頭部MRI画像では、T2強調画像で大脳白質の全般的な淡い高信号像に加えて小脳萎縮、脳梁の菲薄化を認め、さらに基底核の著明な萎縮がないことが共通所見である(図10)。一方T1強調画像では白質高信号を認めることが多く、ある程度の髄鞘化が存在することを示している。経過を追ってもT2で髄鞘化が進展することはなく、T1では逆に髄鞘が消失してくることがある。
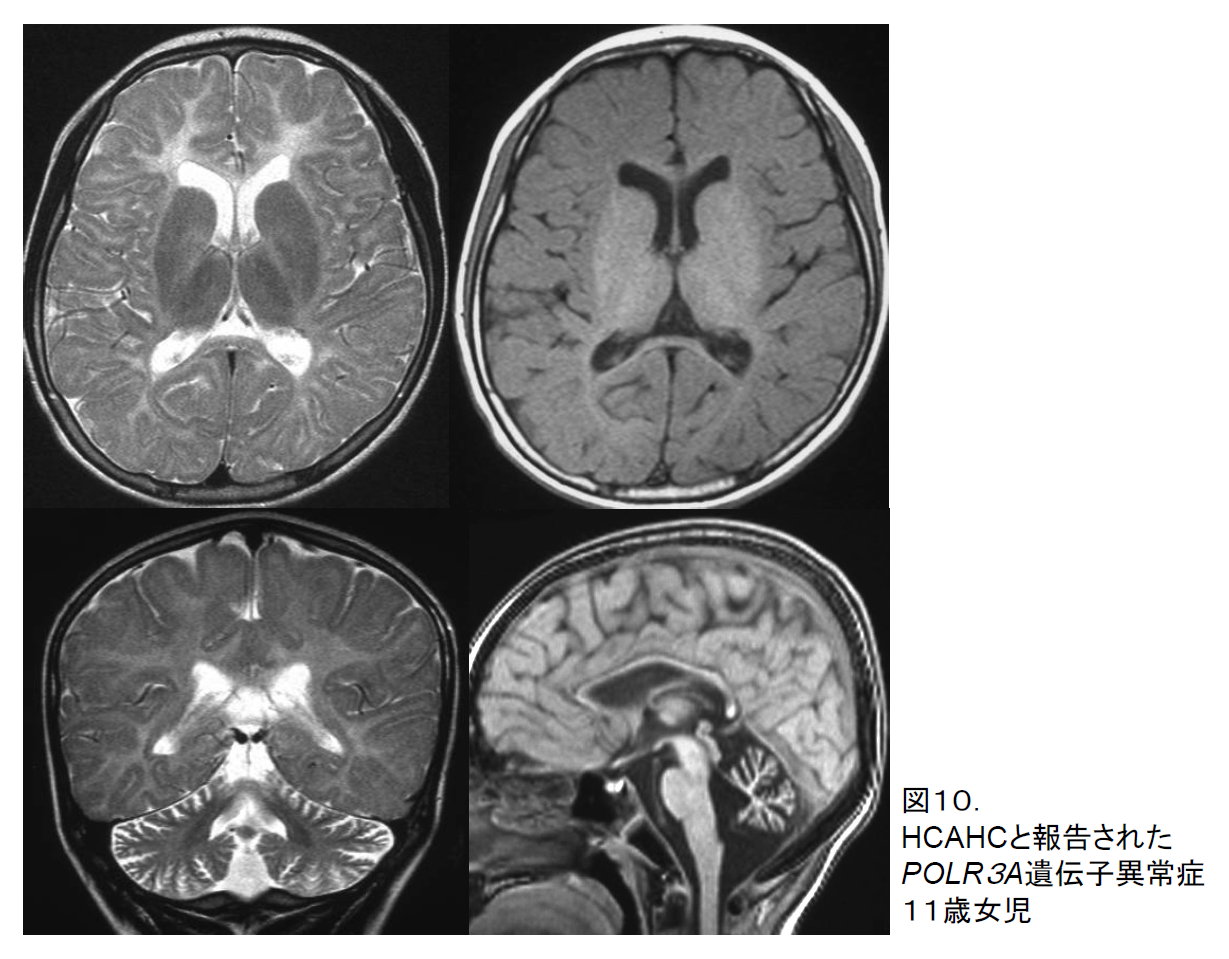
表2.髄鞘低形成に含まれる疾患に特有の症状
認めやすい基本症状:筋緊張低下、痙性四肢麻痺、運動発達遅滞、知的障害(遅滞~退行)、錐体外路症状、眼振、末梢神経障害
| 外表奇形 | 18q-症候群 |
| 乳児期眼振 | PMD、PMD-like |
| 早期ジストニア | H-ABC |
| 先天性白内障 | HCC |
| 小脳失調症状 | POLIII-related leukodystrophy、H-ABC |
| 内分泌異常 | POLIII-related leukodystrophy |
| 歯の異常 | POLIII-related leukodystrophy |
| 近視 | POLIII-related leukodystrophy |
6.文献
- Schiffmann R, van der Knaap MS. An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72:750-759.
- Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010;133:2971-2982.
- Sasaki M, Hanaoka S, Takashima S, et al. MRI and CT findings in Krabbe disease. Pediatr Neurol 1991;7:283-288.
- van der Knaap MS, Boor I, Estévez R. Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in bran ion and water homoeostasis. Lancet Neurol 2012;11:973-985.
- van der Knaap MS, Pronk JC, Sheper GC. Vanishing white matter disease. Lancet Neurol 2006;5:413-423.
- Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet. 2013;92:767-73.
- Miyatake S, Osaka H, Shiina M, et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology 2014;82(24):2230-7.
- Timmons M, Tsokos M, Asab MA, et al. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology 2006;67:2066-2069.
- Sasaki M, Takanashi J, Tada H, et al. Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev 2009;31:582-587.
- Saitsu H, Osaka H, Sasaki M, et al. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am J Hum Genet 2011;89:644-551.
- Bernard G, Chouery F, Putorti ML, et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:415-423.
- Tétreault M, Choquet K, Orcesi S, et al. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am J Hum Genet. 2011;89:652-655.
- Bernard G, Vanderver A. Pol III-Related Leukodystrophies. GeneReviews™ [Internet]. In: Pagon RA, et al. ed. Seattle (WA): University of Washington, Seattle; 1993-2013. 2012 Aug 02.