Wiedemann-Steiner症候群
(Wiedemann-Steiner Syndrome)
[Synonyms: KMT2A関連神経発達障害]
Gene Reviews著者: Sarah E Sheppard, MD, PhD and Fabiola Quintero-Rivera MD.
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2022.5.26. 日本語訳最終更新日: 2022.6.29.
要約
疾患の特徴
Wiedemann-Steiner症候群(WSS)は、発達遅滞、知的障害、特徴的顔貌を主徴とし、これに先天奇形が加わることがある。
顔の特徴としては、側方に広がった太い眉、細く斜下した眼瞼裂、両眼隔離、長い睫毛、広い鼻根、広い鼻尖、薄い上赤唇、豊かな頭髪などがある。
罹患者の約60%に、以前は本症候群だけに現れる特異症候と考えられていた肘の多毛(hypertrichosis cubiti)がみられる。
また、多くの例で、身体の他の場所の多毛が現れる。
その他の臨床症候としては、摂食障害、出生前・出生後の成長障害、癲癇、眼の奇形、先天性心疾患、手の奇形(短指,彎指など)、筋緊張低下、脊椎奇形(特に頸椎の癒合)、腎・子宮奇形、免疫機能不全、脳奇形、歯の形成異常などがある。
診断・検査
発端者におけるWSSの診断は、これを示唆する所見があることに加え、分子遺伝学的検査でKMT2Aの病的バリアントのヘテロ接合を同定することで確定する。
臨床的マネジメント
症状の治療:
体重増加不良や成長障害がみられる罹患者に対しては補助的経管栄養を選択することも含めた摂食機能療法、成長ホルモン分泌不全を示す罹患者に対しては成長ホルモン治療、甲状腺機能低下を示す罹患者に対しては甲状腺ホルモン補充療法、抗体レベルの低下を示す罹患者に対しては免疫グロブリン静脈内投与療法、感染症の頻発する罹患者に対しては抗生剤の予防的投与の検討、大腸機能障害を有する罹患者に対しては便軟化剤もしくは浸透圧性下剤の使用、眼瞼下垂に対しては眼の形成手術、閉塞性睡眠時無呼吸を有する罹患者に対してはCPAP、BiPAPあるいは口蓋扁桃・咽頭扁桃の外科的切除、行動療法、また、癲癇・発達遅滞/知的障害・先天性股関節形成不全・頸椎癒合・眼の奇形・先天性心疾患・腎奇形・子宮奇形・代謝性骨疾患(この場合、ビタミンDの補充が行われることもある)に対しては標準治療を行う。
定期的追跡評価 :
受診ごと:成長パラメーターの計測、栄養状態の評価、便秘の評価、新しい神経症状の出現状況や癲癇発作の活動性に関して、必要に応じ脳波のフォローも含めた評価、脊髄圧迫の臨床徴候に関する評価、不整脈の徴候/症候のモニタリング、発達状況・行動・身体技能に関する評価、感染症の頻発に関するモニタリングを行う。
乳歯萌出以降、6ヵ月ごとに歯科的評価を行う。
小児期には、成長終了ないし初潮確認までの間、早発乳房・原発性無月経に関する評価を行う。
眼科的評価を年に1度、もしくは臨床的必要性に応じて行う。
避けるべき薬剤/環境:
著者らは、抗痙攣薬バルプロ酸の使用により高アンモニア血症をきたしたWSS罹患者を1例経験している。
これはWSS罹患者に特異的な事象ではなく、バルプロ酸の使用にあたっては十分な注意が必要である。
妊娠に関する管理 :
発作性障害を有する妊婦には、妊娠中最も安全な抗痙攣薬を使用していく体制の検討が推奨される。
頸椎奇形により、脊椎の可動域制限や不安定性が惹起される可能性があり、気道管理上、問題になることがある。
胸椎・腰椎領域の奇形や側彎がある場合は、脊髄くも膜下麻酔や硬膜外麻酔の際に問題になることがある。
遺伝カウンセリング
WSSと診断された罹患者で、両親の分子遺伝学的検査が済んでいる例について言うと、その大多数は新生の病的バリアントに起因するものである。
稀に、罹患者である親からの継承によって生じたWSSがみられる。
その状況をみると、どうやらWSSは常染色体顕性遺伝の遺伝様式をとるようである。
したがって、WSS罹患者の子が罹患者となるリスクは50%である。
罹患家系に存在するKMT2Aの病的バリアントの内容が同定されている場合は、出生前・着床前遺伝子検査を行うことが可能となる。
診断
Wiedemann-Steiner症候群(WSS)については、これまでのところ合意済の臨床診断基準というものは公表されていない。
本症候群を示唆する所見
次のような臨床所見、画像所見、家族歴を有する罹患者については、Wiedemann-Steiner症候群を疑うべきである。
臨床症候
- 特徴的顔貌(図1,2,3)
- 肘の多毛(hypertrichosis cubiti)、背中の多毛、下肢の多毛
- 仙骨部皮膚陥凹
- 発達遅滞/知的障害
- 筋緊張低下
- 摂食障害
- 成長障害
- 低身長
- 便秘図

図1:Wiedemann-Steiner症候群の特徴的顔貌を呈する罹患者。
最初の15例については、正貌と側貌の両方を示した。
Sheppardら[2021]より改変。
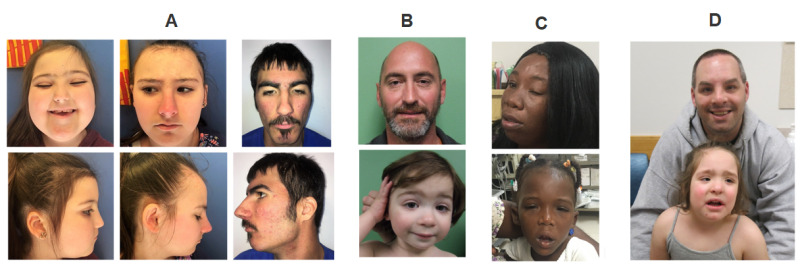
図2:Wiedemann-Steiner症候群の特徴的顔貌を示す罹患者:家族性のもの
A.3同胞例の正貌と側貌
B.父と息子の例
C.母と娘の例
D.父と娘の例
Sheppardら[2021]より改変。

図3:Wiedemann-Steiner症候群の経年変化を示す8例。
Sheppardら[2021]より改変。
画像所見
- 脳のMRIでみられる異常(脳梁の異常と髄鞘形成の異常が最も多い)
- 先天性心疾患
- 腎尿路生殖器系奇形(水腎症を伴う膀胱尿管逆流、男性の停留精巣、女性の子宮欠損が最も多い)
- 椎骨の異常、特に頸椎癒合
家族歴
WSSは、新生の病的変異に起因して生じることが多いため、大多数の発端者は孤発例(家系内で1人だけの発生例)である。
稀に、常染色体顕性遺伝に一致した家族歴(複数世代にわたって男女の罹患者が現れる状態)を示す例がみられる。
診断の確定
発端者におけるWSSの診断は、本症候群を示唆する所見を有することに加え、分子遺伝学的検査によりKMT2Aの病的(もしくはlikely pathogenic)バリアントのヘテロ接合が同定されることにより確定する(表1参照)。
注:(1)ACMGバリアント解釈ガイドラインによると、”pathogenic”と”likely pathogenic”という言葉は臨床現場において同義である。いずれも診断的意義があり、臨床的な意思決定に用いることができる。本章で述べる”病的バリアント”の中にはあらゆるlike pathogenicバリアントも含まれるものとしている。
(2)KMT2Aの意義不明変異のヘテロ接合が検出された場合、それは本疾患の診断確定を意味するものでも本疾患の可能性を否定するものでもない。
(3)KMT2Aはヒストンメチル基転移酵素で、クロマチン仲介性に転写制御を行う働きをする(「分子遺伝学」の項を参照)。
つまり、このタンパク質はゲノムのエピジェネティックな状態を変化させる能力を有しているということになる。
したがって、臨床的重要性の不確かなバリアントについて、その病原性の如何を理解する助けとして、エピジェネティックシグネチャーを評価することも検討対象になりうる。
分子遺伝学的検査の手法としては、遺伝子標的型検査(単一遺伝子検査,マルチ電子パネル)と網羅的ゲノム検査(エクソームシーケンシング,ゲノムシーケンシング)とを、表現型に従って組み合わせて用いることが考えられる。
本症候群を示唆する所見を有しつつも、配列解析やゲノム検査でKMT2Aの病的変異が検出されない例については、エピジェネティックシグネチャーなどのメチル化検査も検討対象になりうる。
遺伝子標的型検査の場合、どの遺伝子の関与が疑われるか、臨床医の側で狙いを定めておく必要がある。
しかし、網羅的ゲノム検査の場合、その必要はない。
「本症候群を示唆する所見」の項で挙げた明確な症候を有する例については、遺伝子標的型検査(手法1)で診断がつく可能性が高い。
しかし、表現型からは、その他数多くある知的障害を伴う遺伝性疾患と区別がつかないような例については、網羅的ゲノム検査(手法2)で診断がつく可能性がより高くなる。
手法1
表現型からWSSの診断が示唆される場合には、分子遺伝学的検査の手法として、単一遺伝子検査ないしマルチ遺伝子パネルが用いられる。
- 単一遺伝子検査
遺伝子内の微小欠失/挿入、ミスセンス、ナンセンス、スプライス部位バリアントを検出するため、最初にKMT2Aの配列解析を実施する。
注:使用する配列解析の手法によっては、単一エクソン・複数エクソン・遺伝子全体といった単位の欠失/重複は検出されない可能性がある。
使用した配列解析の手法で変異が検出されなかった場合は、次のステップとして、エクソンや遺伝子全体の欠失や重複を調べるための遺伝子標的型欠失/重複解析を実施することになる。
- 自閉症/知的障害用マルチ遺伝子パネル
意義不明変異や、現況の表現型と直接つながりのない病的バリアントの検出を極力抑えつつ、疾患の背後にある遺伝的原因の特定に最もつながりやすいと思われるのは、KMT2Aその他の注目すべき遺伝子(「鑑別診断」の項を参照)を含む自閉症/知的障害用マルチ遺伝子パネルである。
注:(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、今このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝子検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
手法2
表現型だけでは、知的障害が現れる他の数多くの疾患と区別ができないといった場合は、網羅的ゲノム検査が検討対象になろう。
網羅的ゲノム検査の場合、どの遺伝子の関与が考えられるかという点を、臨床医の側で検討しておく必要はない。
エクソームシーケンシングが最も広く用いられるが、ゲノムシーケンシングを選択することも可能である。
1塩基単位でみても病的変異が見つからず、それでもWSSの疑いが濃いといった場合は、エピジェネティックシグネチャーなどのメチル化の検査が検討対象になりうる。
網羅的ゲノム検査の基礎的情報についてはここをクリック。
ゲノム検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
表1:Wiedemann-Steiner症候群で用いられる分子遺伝学的検査
| 遺伝子1 | 手法 | その手法で病的変異2が検出される発端者の割合 |
|---|---|---|
| KMT2A | 配列解析3 | 99%近く4 |
| 遺伝子標的型欠失/重複解析5 | 1%未満4 |
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- この遺伝子で検出されているアレルの変異内容に関する情報については、「分子遺伝学」の項を参照のこと。
- 配列解析を行うことで、benign、likely benign、意義不明の変異、likely pathogenic、pathogenic等が検出される。
- Chanら[2019],Sheppardら[2021]
- 遺伝子標的型欠失/重複解析では、遺伝子内の欠失や重複が検出される。
変異の種類としては、遺伝子内の小さな欠失/挿入、ミスセンスバリアント、ナンセンスバリアント、スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。
配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失/重複の検出を目的に設計された遺伝子標的型マイクロアレイなど、さまざまなものがある。
遺伝子標的型欠失/重複解析では、1エクソンから当該遺伝子全体に至るまでの欠失が検出されるものの、大きな欠失の切断点、ないし隣接する遺伝子群の欠失については、こうした手法では検出されない場合がある。
臨床的特徴
臨床像
Wiedemann-Steiner症候群(WSS)は、発達遅滞、知的障害、特徴的顔貌を主徴とし、これに他の先天奇形が伴う例もみられる。
KMT2Aの病的バリアントの同定された例が、現在までに200人以上、医学文献に報告されている[Jonesら2012,Mendelsohnら2014,Stromら2014,Bramswigら2015,Calvelら2015,Steelら2015,Yuanら2015,Miyakeら2016,Stellacciら2016,Aggarwalら2017,Bogaertら2017,Enokizonoら2017,Min Koら2017,Sunら2017,Baerら2018,Lebrunら2018,Liら2018,Stoyleら2018,Chanら2019,Chenら2019,Feldmanら2019,Negriら2019,Giangiobbeら2020,Matisら2020,Mendoza 2020,Demirら2021,Di Fedeら2021,Nardelloら2021,Sheppardら2021]。
これらの報告に基づいて、以下に、本疾患で現れる表現型の特徴について述べる。
表2:Wiedemann-Steiner症候群:代表的症候の出現頻度
| 症候 | その症候を有する割合 | コメント |
|---|---|---|
| 発達遅滞/知的障害 | 97% | |
| 特徴的形態異常 | 75% | 多くみられるのは、太い眉、長い睫毛、両眼隔離、細く斜下した眼瞼裂。 |
| 筋緊張低下 | 63% | |
| 便秘 | 50% | |
| 成長障害 | 64% | |
| 摂食障害 | 62% | ふつう、乳児期や小児期のみ。 |
| 耳鼻咽喉科的問題 | 63% | Sheppardら[2021]のコホート中60%が耳鼻咽喉科的問題をもっていた。 他のコホートでも、睡眠時無呼吸(18%)、難聴(3%)、粘膜下口蓋裂(3%)などの問題が報告されている。 |
| 多毛症 | 44%-75% | 最も多くみられるのは顔(太い眉[73%],長い睫毛[75%])で、次いで背中(69%)、肘(58%)、下肢(44%)。 |
| 低身長1 | 57%以内 | |
| 画像上の脳の異常 | 49%以内 | 脳梁の異常と髄鞘形成の異常が最も多い。 |
| 歯の異常2 | 57% | 乳歯の早期脱落と永久歯の早期萌出を伴う歯牙年齢の亢進。 |
| 仙骨部の奇形 | 45%以内 | 最も多くみられるのは仙骨部皮膚陥凹、次いで脊髄係留、潜在性二分脊椎。 |
| 脊椎奇形 | 46% | 最も多くみられるのは頸椎癒合。 |
| 腎尿路生殖器系奇形 | 46.8%以内 | 腎奇形(29%),その他に子宮、精巣、外性器の奇形もみられる。 |
| 精神神経系の問題 | 17%-38% | 多動(38%),攻撃的行動(28%),自閉症スペクトラム障害(17%) |
| 眼の異常 | 18%-32% | 最も多くみられるのは斜視、次いで乱視や眼瞼下垂。 |
| 心臓の異常 | 29% | 最も多くみられるのは動脈管開存と心室中隔欠損。 |
| 内分泌系の問題 | 30%-64% | 成長ホルモン分泌不全(30%),副腎皮質性思春期早発(27%),MRIでみられる下垂体の異常(64%),骨年齢の異常(63%) |
| 癲癇発作 | 18% | 焦点性発作,欠神発作,全般発作 |
| 免疫系の問題 | 21%-54% | 免疫グロブリン値の異常,肺炎球菌ワクチンの反応性低下,反復性感染症(21%) |
- ここでいう低身長とは、身長が年齢性別ごとの標準に対し-2SD未満ないし5パーセンタイル未満、もしくは生後の成長不全をいう。
- Aggarwalら[2017],Sheppardら[2021]
特徴的形態異常
肘の多毛については、以前は本症候群だけに現れる特異症候と考えられていたものであるが、これが罹患者の大多数にみられる[Stromら2014,Aggarwalら2017,Baerら2018,Sheppardら2021]。
特徴的顔面症候としては、次のようなものが挙げられる。
- 太い眉
- 長い睫毛
- 細い眼瞼裂
- 両眼隔離
- 幅広の鼻根,広く丸まった鼻尖
- 側方(ないし他の方向)へ広がった眉
- 眼瞼裂斜下
- 眼瞼下垂
- 目立つ二重弓形(口唇)
- 薄い上口唇
- 後方に傾いた耳
発達遅滞
罹患者の大多数には発達指標に遅れがみられ、一部、発語や歩行が生じないままになってしまう例もみられる[Baerら2018,Liら2018,Chanら2019,Sheppardら2021]。
発達指標の中央値は以下の通りである。
- お座り…10ヵ月(範囲は6-36ヵ月)
- 立ち上がり…17ヵ月(範囲は8-60ヵ月)
- 歩行…20ヵ月(範囲は11-60ヵ月)
- 初語…18ヵ月(範囲は8-60ヵ月)
知的障害と学業
罹患者の多くが、ある程度の支援教育を必要とするものの、ある研究によると、成人罹患者の大多数は高校を卒業していたという。
- 一部の罹患者は、社会人として仕事を維持できるだけの能力を有する。
- 検査した範囲では、IQは40から85(中央値65)である。
- 発達ならびに学習の問題に関するより詳細な情報については、Feldmanら[2019]ならびにSheppardら[2021]を参照されたい。
行動の問題
罹患者の約30%が、自傷のような攻撃的行動を有する一方、中には他者への身体的攻撃や癇癪を示す例もみられる。
罹患者の約20%は、自閉症スペクトラム障害を窺わせるような自閉兆候を有する。
WSSの子どもは、家族から、優しく楽しい性格と評される例が多い[Sheppardら2021]。
成長
大多数の罹患者は、出生時体重が25パーセンタイルを下回る。
- 罹患者の3分の1は、体重が各年齢の5パーセンタイルを下回るレベルで推移する。
- 罹患者の60%近くが低身長を示す(ここで言う低身長とは、その年齢・性別において5パーセンタイル未満、その年齢・性別において-2SDを下回る、「出生後の成長障害」のいずれかを満たすものを指す)。
骨年齢評価用のX線写真では、65%近くが異常(遅延,亢進,不均衡 ― 成熟レベルの異なる骨が混在する状態)を示した。
- 罹患者の約3分の1は、小頭症(頭囲が、当該年齢の5パーセンタイル未満([訳注:原文は「5パーセンタイル超」となっているが、誤りと思われる]、もしくは、当該年齢の平均値の-2SDを下回る)を有する。
消化器系
罹患者の3分の2に成長障害(FTT)と摂食障害の既往がみられる。
ただ、経管栄養を要するのは4分の1未満である。
FTTは一時的なものなので、暫間的対応として経鼻胃管が有用な場合もある。
便秘は、罹患者の約半数にみられ、FTTを伴う罹患者により多くみられる。
神経系
- 仙骨部皮膚陥凹を筆頭とする仙骨部の奇形が、罹患者の約45%にみられる。
- その他の仙骨部の奇形には、潜在性二分脊椎や脊髄係留(これについては手術を要する)などがある。
- 筋緊張低下は、罹患者の約3分の2にみられる。
- 筋緊張低下は出生の段階ですでにみられることがあり、その場合、胃瘻造設や経鼻胃管が勧められるような事態に至る可能性もある。
- 罹患者の中には、胃瘻経由で補助栄養を継続する必要があるような例もみられる。
- 中には経時的に筋緊張低下が改善していくような例もみられる。
- 癲癇発作が、罹患者の5分の1近くにみられる。
- 癲癇発作の種類としては、欠神発作、複雑部分発作、眼瞼ミオクロニー、強直間代発作、熱性けいれん、点頭癲癇などがある[Helbigら2016]。
- 癲癇性脳症の報告がみられる。
- 治療に関する情報は少ないものの、多剤耐性の報告がみられる。
- 中枢性無呼吸の報告もみられる。
また、一部に治療を要しない罹患者がみられた一方で、ラモトリギンを用いた治療が有効であったとの報告がみられる[Koenigら2010,Stellacciら2016,Baerら2018,Sheppardら2021]。
脳のMRI所見
脳の画像診断を受けた罹患者の約半数に、脳の器質的異常が認められている。
具体的所見としては、以下のようなものがある[Baerら2018,Sheppardら2021]。
- 脳梁の異常
- 髄鞘形成の異常
- 高信号の白質内点状病巣、白質体積の減少を示す像、白質形成不全といった白質の変化
- 比較的狭小な大孔、扁平頭蓋底といったChiariⅠ型奇形スペクトラム
- 脳室周囲結節状異所性灰白質
- 脈絡叢嚢胞
- 下垂体後葉欠損、トルコ鞍形態異常、視床下部下垂体系の低形成を伴う後葉の異所性高輝度点、下垂体低形成などの下垂体関連の異常(このセクション内の「内分泌系」の項を参照)
- 前頭葉両側の多小脳回をはじめとする皮質奇形[Grangeiaら2020,Nardelloら2021]。
- 視神経の低形成[Chenら2019]
- 中脳水道狭窄や第三脳室拡張などの脳脊髄液の異常[Aroraら2020]
- 脳萎縮
- 小脳虫部低形成[Di Fedeら2021]
- 小脳萎縮[Giangiobbeら2020]
皮膚/毛髪
多毛症が多く出現し、罹患者の約50%-75%にみられる。
具体的には、次のような形で現れる。
- 眉
- 長い睫毛
- 豊かな頭髪
- 背中の多毛症
- 肘の多毛症
- 下肢の多毛症
骨格/四肢
椎骨奇形は、筋骨格系の症候として最も多くみられるもので、罹患者の約半数にこれがみられる。
椎骨奇形の大半は頸椎癒合で、その中で最も多いのはC2-C3の癒合である。
ただ、中には、胸椎、腰椎、仙椎に異常が現れる例もみられる。
その他の骨格奇形としては次のようなものがある。
- 肋骨奇形(例えば、数の減少、低形成、頸肋)が罹患者の約3分の1にみられる。
- 幅広の第1指、ないし先細りの第2-5指が、罹患者の約4分の1にみられ、一部に胎児期の指腹隆起の遺残がみられる例もみられる[Miyakeら2016,Enokizonoら2017,Wangら2021]。
- 罹患者の5分の1未満ながら、脊柱側彎を示す例がみられ、稀に、手術を要するほど重度のものもみられる。
- 少数の罹患者で、漏斗胸が報告されている。
- 股関節形成不全も少数の罹患者で報告されているが、骨盤位分娩に伴ってこれが現れた例はみられない。
手術を要した例が数例、Pavlikハーネスを要した例が1例報告されている。
- 舟状頭蓋が1例、三角頭蓋が1例報告されている[Nardelloら2021]。
著者ら自身も、もう1例、三角頭蓋の例を経験している。
心臓
心臓の評価を受けた例に限っていうと、約3分の1に心臓の異常が認められている。
具体的には以下の通りである。
- 器質的な心奇形
動脈管開存,卵円孔開存,右大動脈弓,大動脈弁閉鎖不全,大動脈二尖弁,心房中隔欠損,心室中隔欠損,Fallot四徴,迷走右鎖骨下動脈,僧帽弁逸脱,右胸心,大動脈弁肥厚,僧帽弁逆流,三尖弁逆流,大動脈騎乗,大動脈弁肥厚(訳注:「大動脈弁肥厚」が重複しており、何らかの編集上の誤りがあるものと思われる)
- 第3度房室ブロックを含む不整脈(ペースメーカーを要した1例あり)[Bogaertら2017,Liら2018]。
- 肺高血圧
- 失神発作
腎尿路生殖器系
腎尿路生殖器系の異常が、罹患者の半数近くにみられる。
- 腎奇形がおおむね4分の1にみられる。
具体的には、水腎症を伴う膀胱尿管逆流などがある。
- 子宮ないし精巣の奇形が20%近くにみられる。
具体的には、女性では子宮欠損、男性では停留精巣などがある。
- 約10%に外性器の奇形がみられる。
具体的には、突出した陰核、低発達の陰嚢、尿道下裂などがある。
眼
以下のような、さまざまな眼の異常がみられる。
- 斜視
- 乱視
- 遠視
- 近視
- 弱視
- 鼻涙管の異常
- 眼瞼下垂
- 稀に、白内障,コロボーマ,緑内障
耳鼻咽喉系
閉塞性睡眠時無呼吸が最も多くみられる症候で、罹患者のおおむね4分の1にみられる。
睡眠時無呼吸を呈する例のほぼ20%で、口蓋扁桃摘出術やアデノイド切除術が必要となる。
歯/口腔
罹患者の半数超が歯の問題を有する。
最も多くみられるのは、乳歯の早期喪失、永久歯の異常に早い萌出という形で現れる歯牙年齢の亢進である。
その他の症候としては、咬合異常、歯の形態異常、無歯症、過剰歯、エナメル質形成不全、齲蝕、急速拡大装置を必要とするような高口蓋、歯肉の問題等がある。
内分泌系
内分泌系の問題としては、次のようなものがある。
- 低身長(このセクション内の「成長」の項を参照)
- 思春期早発症
- 月経過多,多嚢胞性卵巣症候群,月経不順
- 脳のMRIでみられる下垂体の異常(このセクション内の「脳のMRI所見」の項を参照)
- 血清成長ホルモン(GH)値やインスリン様成長因子1(IGF-1)値の低下、ないし成長ホルモン分泌刺激試験で明らかになる成長ホルモン分泌不全
- 骨減少症
- 甲状腺機能低下(先天性甲状腺機能低下や慢性甲状腺炎など)
- 副甲状腺機能低下
免疫系
免疫系の問題の報告もみられる。
具体的には、以下のようなものがある[Jonesら2012,Stellacciら2016,Bogaertら2017,Baerら2018,Sheppardら2021]。
- 分類不能型免疫不全症などの免疫不全
- ワクチンに対する反応性低下
- 反復性感染症の既往
- 反復性の不明熱
- 好酸球増多症[Zhangら2019]
反復性の肺感染症を示した1男児が、敗血症により死亡している[Bramswigら2015]。
予後
WSS罹患者の寿命が一般集団と異なるかどうかという点については、よくわかっていない。
文献で、複数の成人罹患者が報告されている[Liら2018,Feldmanら2019,Sheppardら2021]。
就業していない成人が何人かいる一方で、昼間のリハビリに通っている人が1人、建設業を営んでいる人が1人、特別な勉学上の支援を受けることなく大学に通っている人が1人といった状況である。
WSSは、これを有する成人から子へ、継承される可能性のある疾患である。
ところが、障害を有する成人の多くは、高度な遺伝学的検査を受けていない。
そのため、本疾患を有する成人は実際より少数しか把握されておらず、報告数も少数にとどまっていることが考えられる。
遺伝型-表現型相関
いくつかの遺伝型-表現型相関が明らかになっている。
- KMT2Aの機能喪失型変異を有する罹患者のほうが、これを有しない罹患者より、筋緊張低下を示すことが多い[Sheppardら2021]。
- 一方、機能喪失型変異以外の変異を有する罹患者には、癲癇発作を示す例が多い[Sheppardら2021]。
- CXXC DNA結合ドメイン(アミノ酸1147-1195)のミスセンスバリアントを有する例は、神経発達に、より重大な問題を有する可能性がある[Min Koら2017,Liら2018,Lebrunら2018]。
命名法について
1970年、Dr P Beightonが、家族性の肘多毛症(「多毛肘」症候群;“hairly elbows” syndrome)を有する父と2児の例を報告した[Beighton 1970]。
そしてその後、Dr HR Wiedemannらが、成長障害、発達遅延、斜視、腎杯拡張、特徴的顔貌を呈する1男性例を報告した[Wiedemannら1989]。
2000年、Dr CE SteinerとDr AP Marquesが、BeightonやWiedemannの報告した症例に類似した症候を示す1女性例を報告し、これらが同一の疾患である可能性について言及した[Steiner & Marques 2000]。
これまで、WSSの表現に「多毛肘症候群」を用いた文献がみられはするものの、現在では、「Wiedemann-Steiner症候群」が広く用いられている。
あるいは、Bieseckerら[2021]の提唱した命名法に従う形で、「KMT2A関連神経発達障害」と呼ぶのも良いかもしれない。
発生頻度
WSSの発生頻度は不明である。
文献では、250例近くの報告が存在する。
成人罹患者の中には、自分の子どもがWSSの評価を受ける一環として検査を受けた結果、自身もWSSであると診断されるような例が複数みられる。
したがって、WSSという診断がなされていない隠れた罹患者が、実際は多く存在する可能性が考えられる[Sheppardら2021]。
遺伝子の上で関連のある疾患(同一アレル疾患)
KMT2Aの生殖細胞系列の病的変異に関しては、このGeneReviewで述べた以外の表現型は知られていない。
孤発性の腫瘍(急性白血病,胸腺腫,乳癌など)で、それ以外のWiedemann-Steiner症候群の症候を一切有しない例において、その腫瘍組織中に、生殖細胞系列の変異としては現れることのないKMT2Aの病的バリアント(あるいはKMT2Aを含むゲノム再編成)がしばしば確認される(www.mycancergenome.org)。
こうした場合、腫瘍発生素因は非遺伝性である。
より詳細な情報については、「癌ならびに良性腫瘍」の項を参照されたい。
鑑別診断
Wiedemann-Steiner症候群(WSS)と症候が重なる疾患は、多岐にわたる。
表3は、WSSと一致する臨床症候を有する例において、最も多く検討がなされる疾患群をまとめたものである。
鑑別の対象として検討の対象になる頻度がこれらより低い疾患群については、Sheppardら[2021]を参照されたい。
表3:Wiedemann-Steiner症候群との鑑別を要する主要疾患
| 遺伝子 | 鑑別すべき疾患 | 遺伝様式 | 鑑別すべき疾患でみられる臨床症候 | |
|---|---|---|---|---|
| WSSと重なる症候 | WSSと異なる症候 | |||
| ARID1A ARID1B ARID2 DPF2 SMARCAA4 SMARCB1 SMARCC2 SMARCE1 SOX4 SOX11 |
Coffin-Siris症候群 | AD | 症候群性低身長 長い睫毛,目立つ眉を伴う多毛症 |
遠位指趾低形成(通常、第5指趾) |
| BRAF KRAS LZTR1 MAP2K1 NRAS PTPN11 RAF1 RIT1 SOS1 |
Noonan症候群 | AD AR1 |
低身長,心臓の器質的異常,肺高血圧,眼の異常,筋緊張低下 | 先天性心疾患(50-80%)がより高頻度に現れるほか、肺動脈弁狭窄もみられるが、多毛症はみられない。 |
| BRD4 HDAC8 NIPBL RAD21 SMC1A SMC3> |
Cornelia de Lange症候群 | AD XL2 |
多毛症,低身長,成長障害,発達遅滞/知的障害,眼の異常,反復性感染症 | 四肢の症候がみられ、上肢の欠損は、前腕の完全欠損といった重度の欠損から、種々のタイプの乏指に至るまで、幅がみられる。 |
| CREBBP EP300 |
Rubinstein-Taybi症候群 | AD> | 低身長,発達遅滞/知的障害,多毛症,先天性心疾患 | 高インスリン症,顔をしかめたような特徴的スマイル |
| KDM6A KMT2D |
Kabuki症候群 | AD XL3 |
多毛症,低身長,成長障害,発達遅滞/知的障害,多発奇形 | 高インスリン症 |
| TASP1 | Suleiman- El-Hattab症候群 (OMIM 618950) |
AR | 発達遅滞,小頭症,成長障害(FTT)を伴う摂食障害,反復性呼吸器感染症,心血管系奇形,楽しい性格,特徴的顔貌4 | 眉間の突出 |
AD=常染色体顕性;AR=常染色体潜性;XL=X連鎖性
- Noonan症候群(NS)は、大多数が常染色体顕性の遺伝様式を示す。
ただ、LZTR-1関連NSだけは、常染色体顕性ないし常染色体潜性の遺伝様式をとる。
- NIPBL関連Cornelia de Lange症候群(CdLS)、RAD21関連CdLS、SMC3関連CdLS、BRD4関連CdLSは常染色体顕性の遺伝様式をとる。
- KMT2D関連Kabuki症候群(KS)は常染色体顕性の遺伝様式をとる。
- TASP1はKMT2Aの切断に関与する。
HDAC8関連CdLS、SMC1A関連CdLSについては、X連鎖性の遺伝様式を示す。
KDM6A関連KSについては、X連鎖性の遺伝様式を示す。
WSSとSuleiman-El-Hattab症候群との間の類似性に、このことが関与している可能性が考えられる[Sheppardら2021]。
(訳注:この注4で、TASP1とKMT2Aはともにイタリック体表記になっているが、ここではタンパク質の有する機能について述べているように思われるので、イタリック体にしないのが正しいのではないかと思われる)
臨床的マネジメント
Wiedemann-Steiner症候群(WSS)の治療のガイドラインが公表されている。
Baerら[2018](全文はこちら)、Sheppardら[2021] (全文はこちら)を参照されたい。
注:参照には、機関からのアクセス、もしくは購入が必要である。
最初の診断に続いて行う評価
WSSと診断された罹患者については、疾患の範囲やニーズを把握するため、診断に至る過程における評価の一部としてすでに実施済ということでないようなら、表4にまとめたような評価を行うことが推奨される。
表4:Wiedemann-Steiner症候群罹患者に対し、最初の診断後に行うことが推奨される評価
| 系/懸念事項 | 評価項目 | コメント |
|---|---|---|
| 体格 | 成長パラメーターの計測 | FTTや、成長ホルモン分泌不全を含む成長不良の罹患者を同定することを目的として行う。 |
| 神経系 | 神経学的評価 |
|
| 発達 | 発達評価 |
|
| 精神/行動 | 精神神経学的評価 | 生後12ヵ月超の罹患者について:睡眠障害、攻撃的行動、自閉症スペクトラム障害を窺わせる徴候等の行動上の懸念に関するスクリーニングを行う。 |
| 筋骨格系 | 脊椎のX線写真、脊椎のCT、トノデンソメトリーを検討する。 | 管理に役立てることを目的として行う。 |
| 整形外科/物療医学/リハビリテーション医学/理学療法・作業療法的評価 | 以下の点の評価を含めて行う。 粗大運動技能と微細運動技能
|
|
| 消化器系/摂食 | 消化器/栄養/摂食チームによる評価 |
|
| 便秘の徴候/症候に関する評価 | ||
| 眼 | 眼科的評価 | 斜視、眼瞼下垂、屈折異常、その他の稀な合併症の診断・治療を目的とする |
| 耳鼻咽喉/口腔 | 睡眠検査を検討 | 睡眠時無呼吸の評価 |
| 1歳超の罹患者については、小児歯科的評価 | 乳歯の早期脱落、永久歯の早期萌出に関する評価 | |
| 心血管系 | 心エコーと心電図 | 先天性心疾患の有無の評価、不整脈のスクリーニングを目的として行う。 |
| 腎尿路生殖器系 | 腹部超音波検査 | 腎や膀胱の異常の評価を目的として行う。 |
| 女性の新生児あるいは思春期以降の女性については、体外式骨盤超音波検査を検討する1。 | 子宮奇形のスクリーニングを目的として行う。 | |
| 皮膚 | 皮膚科的評価 | 多毛症の管理上の補助として、家族から要望があった場合に行う。 |
| 内分泌系 | 内分泌内科的評価 |
|
| 免疫 | 免疫学的評価 |
|
| 遺伝カウンセリング | 遺伝専門職2の手で行う。 | 罹患者やその家族に対し、WSSの本質、遺伝様式、そのもつ意味に関する情報提供を行うことで、医学的、個人的決断に資することを目的として行う。 |
| 家族の支援,情報提供源 | 下記のニーズに関する評価を行う。
|
- 直近にエストロゲン補充を受けていない女性については、骨盤の超音波検査、あるいは骨盤のMRIを行ってさえ、子宮の確認が十分に行えないことがある。
こうした状況下においては、両手法とも、子宮欠損の確認用としては不十分である。
評価のための婦人科への紹介も検討に値するかもしれない。 - 臨床遺伝医、認定遺伝カウンセラー、認定上級遺伝看護師をいう。
- WSS罹患者の親に特化した情報提供源としては、WSS Foundation(「情報提供源」の項を参照)やWSS のFacebookのグループなどがある。
症状に対する治療
表5:Wiedemann-Steiner症候群罹患者の症候に対する治療
| 症候/懸念事項 | 治療 | 考慮事項/その他 |
|---|---|---|
| 癲癇 | 経験豊富な神経内科医あるいは癲癇専門医による抗痙攣薬を用いた標準治療 |
|
| 発達遅滞/知的障害/行動の問題 | 「発達遅滞/知的障害の管理に関する事項」の項を参照。 | |
| 先天性股関節形成不全 | 整形外科医による標準治療 | Pavlikハーネスの使用、重度例では外科的介入などが考えられる。 |
| 頸椎癒合 | 整形外科医による個別的評価 | 稀ながら、椎弓切除術を要する罹患者がみられる。 |
| 体重増加不良/成長障害 |
|
臨床的摂食評価をためらわずに行うことが必要。 |
| 腸の機能障害 | 必要に応じ、便軟化剤、腸運動促進薬、浸透圧性薬剤、瀉下薬の使用。 | |
| 斜視,乱視,その他の屈折異常 | 眼科医による標準治療 | |
| 眼瞼下垂 | 眼の形成手術を検討する。 | |
| 睡眠時無呼吸 | CPAP、BiPAP、あるいは口蓋扁桃・咽頭扁桃の外科的切除などが考えられる。 | |
| 先天性心疾患と不整脈 | 心臓病専門医による標準治療 | 抗不整脈薬ないし外科的手法による治療が考えられる。 |
| 腎奇形 | 腎臓内科医による標準治療 | |
| 子宮奇形 | 婦人科医による標準治療 | |
| 成長ホルモン分泌不全 | 成長ホルモン治療 | この治療は、通常、内分泌内科医の手で行われる。 |
| 甲状腺機能障害 | 甲状腺ホルモン補充療法 | |
| 代謝性骨疾患 | 内分泌内科医による標準治療 | ビタミンD補給が考えられる。 |
| 感染症の頻発/免疫機能不全 | 抗体値の低い例については免疫グロブリン静脈内投与、感染症が頻発する例については予防的な抗生剤投与を検討する。 | 免疫医による管理が推奨される。 |
| 家族/地域社会 | ・家族が地域の情報提供源とつながり、息抜きや支援が得られるよう、ソーシャルワーカーが適切な形で関与できるようにする。 ・多分野にまたがる受診予約、機器使用、服薬、物品提供のやりくりをつけるための調整 |
|
- 代表的な癲癇発作に関し、親や介護者に対し教育を行うことが適切である。
癲癇と診断された子どもに対する非医学的介入ならびに対処法に関する情報については、「Epilepsy & My Child Toolkit」を参照されたい。
発達遅滞/知的障害の管理に関する事項
アメリカでは発達遅滞者、知的障害者の管理に向けた推奨として、次のような内容のものがある。
ただ、そうした標準的推奨事項は、国ごとに異なったものになることもあろう。
0-3歳
作業療法、理学療法、言語治療、摂食治療、乳児精神保健サービス、特別支援教育、感覚障害教育が受けられるよう、早期介入プログラムへの紹介が望ましい。
早期介入プログラムは、アメリカでは連邦政府が費用を負担して行われる制度で、すべての州で利用可能である。
このプログラムは、個人個人の治療上のニーズに合わせた在宅サービスを提供するものである。
3-5歳
アメリカでは、地域の公立学区(訳注:ここで言う「学区」というのは、地理的な範囲を指す言葉ではなく、教育行政単位を指す言葉である)を通じて発達保育園に入ることが推奨される。
入園前には必要なサービスや治療の内容を決定するための評価が行われるとともに、運動、言語、社会性、認知の遅延を基礎として認定された子どもに対しては、個別教育計画(IEP)の作成が行われる。
通常、早期介入プログラムが支援する形で、こちらへの移行が進められる。
発達保育園は独立運営である。
医学的問題から通園できない子どもに対しては、在宅サービスが提供される。
全年齢
各地域、州、アメリカの連邦教育行政機関が適切な形で関与できるよう、そして、良好な生活の質を最大限確保する支援を親に対してできるよう、発達小児科医への紹介が推奨される。
頭に入れておくべき点は以下の通りである。
- 個別教育計画(IEP)サービスについて
- 認定を受けた子どもに対して、IEPを通じて、個別の指導や関連サービスが提供される。
- IEPサービスについては、何らかの変更を要するかどうか、毎年見直しが行われる。
- 特別支援教育法の定めに従い、IEPの策定された子どもは、学校現場で実現可能な範囲で最も制約の少ない環境下に置かれ、時間と場所の許す範囲で、可能な限り多くの一般教育を受けられるようにする。
- IEPのチームには、必ず視覚・聴覚の支援員が加わる形で、子どもが学習教材を使用できるよう支援していくことになっている。
- 理学療法、作業療法、言語治療については、子どもが学習教材を使用していく上で障害があるようであれば、IEPの中に組み込んでいくことになる。
この範囲を超えるサービスについては、罹患者の要求に従って、私費負担での支援治療が検討されることになろう。
個人個人に推奨される治療の具体的内容については、必要に応じて発達小児科医が決めていくことになろう。
- 子どもがティーン世代に達したら、後々の移行計画についての話し合いを行い、そこで決められた内容をIEPに組み込んでいく必要がある。
IEPのサービスを受けている人に対して、公立学区は21歳までサービスを提供する決まりになっている。
- 504プランについて
教室内で前のほうへの着席、補助機器の使用、代書者の起用、業間の休み時間の延長、課題の変更、大きい教科書の使用といった配慮や変更が必要な人に対しては、504プラン(第504項:障害に基づく差別を禁じたアメリカの連邦法)が検討される。
- 発達障害者福祉局(DDA)への登録について
発達障害者福祉局(DDA)への登録が推奨される。
DDAは、アメリカの公的機関で、認定を受けた人に対してサービスや支援を提供している。
認定基準は州によって異なるものの、ふつうは、診断名やそれに伴う認知/適応障害の度合に従って決定がなされる。
- 補足的所得補償制度(SSI)について
収入が少なく、情報も不足している家族については、障害児のための補足的所得補償制度の認定を受ける道がある。
運動機能障害
粗大運動機能障害
- 可動性を最大限確保するため、ならびに、遅発性の整形外科的合併症(例えば、脊柱側彎や股関節脱臼)のリスクを低減させるため、理学療法が推奨される[Mendoza 2020]。
- 必要に応じて、耐久性医療機器や体位保持機器(例えば、車椅子、歩行器、バスチェア、補装具、障害者用ベビーカー)の使用を検討する。
微細運動機能障害
摂食、身だしなみ、着替え、筆記などの機能に問題が生じる微細運動技能の障害に関しては、作業療法が推奨される。
口腔運動機能障害
口腔運動機能障害の評価は、来院ごとに行う必要がある。
また、摂食哺乳時の窒息/嘔吐、体重増加不良、呼吸器疾患の頻発、特別な原因が見出せない摂食拒否といった状況がみられる場合は、X線による嚥下検査を行う必要がある。
食物の経口摂取を安全に行えることが前提ではあるが、協調ないし感覚に関連する摂食の問題を改善していくための摂食治療(通常、作業療法士か言語治療士の手で行われる)が推奨される。
安全のため、食餌にとろみをつけたり冷やしたりすることもある。
摂食機能障害が重度である場合は、経鼻胃管や胃瘻管が必要になるようなこともある。
コミュニケーションの問題
表出言語に障害をもつ罹患者に対しては、それに代わるコミュニケーション手段(例えば、拡大代替コミュニケーション[AAC])に向けての評価を検討する。
AACに向けた評価は、その分野を専門とする言語療法士の手で行うことが可能である。
この評価は、認知能力や感覚障害の状況を考慮に入れながら、最も適切なコミュニケーションの形を決めていこうというものである。
AACの手段としては、絵カード交換式コミュニケーションシステムのようなローテクのものから、音声発生装置のようなハイテクのものまで、さまざまなものがある。
一般に信じられていることとは反対に、AACはスピーチの発達を妨げるようなものではなく、むしろ理想的な言語発達に向けた支援を与えてくれるものである。
社会/行動上の懸念事項
小児に対しては、応用行動分析(ABA)をはじめとする自閉症スペクトラム障害の治療で用いられる治療的介入の導入に向けた評価を行うとともに、実際にそれを施行することがある。
ABA療法は、個々の子どもの行動上の強みと弱み、社会性に関する強みと弱み、適応性に関する強みと弱みに焦点を当てたもので、ふつう、行動分析に関する学会認定士との1対1の場で行われる。
発達小児科医を受診することで、両親に対し、適切な行動管理の指針を指導したり、必要に応じ、注意欠陥多動性障害に用いられる薬剤をはじめとする処方を行ったりといったことが可能になる利点がある。
深刻な攻撃的、破壊的行動に関して懸念があるときは、小児精神科医への相談という形での対応が考えられる。
定期的追跡評価
表6:Wiedemann-Steiner症候群罹患者に推奨される定期的追跡評価
| 系/懸念事項 | 評価法 | 実施頻度 |
|---|---|---|
| 成長/摂食 |
|
来院ごと2 |
| 消化器系 | 便秘に関するモニタリング | |
| 神経系< |
|
|
| 発達 | 発達の進み具合や教育上のニーズに関するモニタリング | |
| 精神/行動 | 注意力や攻撃的行動に関する行動学的評価 | |
| 筋骨格系 | 運動能力や自助能力に関する物療医学的、作業/理学療法的評価 | |
| 心血管系 | 不整脈の徴候/症候に関するモニタリング | |
| 免疫系 | 感染症頻発の徴候に関するモニタリング | |
| 家族/地域社会 | 家族の感じているソーシャルワーカーの支援(例えば、緩和や息抜きのためのケア、在宅看護、地域の情報提供源など)の必要性や、協調のとれた管理に向けての必要性についての評価 | |
| 耳鼻咽喉/口腔 | 睡眠障害の徴候/症候に関するモニタリング | |
| 歯科的評価 | 乳歯萌出後、6ヵ月ごと | |
| 内分泌系 | 早発乳房や思春期異常(原発性無月経)などに関する評価 | 小児期に開始し、成長終了/初潮まで |
| 眼 | 眼科的評価 | 年に1度もしくは臨床的必要性に応じて |
- 必要に応じ、脳波のフォローも行う。
- 受診頻度は個々のニーズによって異なるものの、一般的に言うと、幼児期には6ヵ月ごと、その後、学齢期には年に1度へと移行する。
避けるべき薬剤/環境
著者らは、抗痙攣薬バルプロ酸の使用により高アンモニア血症をきたしたWSS罹患者を1例経験している。
これはWSS罹患者に特異的な事象ではなく、バルプロ酸の使用にあたっては十分な注意が必要である。
リスクを有する血縁者の評価
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
妊娠に関する管理
WSSの女性の中には、発作性障害をもち、抗痙攣薬で治療が行われている例がみられる。
一般に、癲癇ないし発作性障害(原因を問わず)を有する女性は、発作性障害を有しない妊婦に比べ、妊娠中の死亡リスクがより高くなる。
妊娠中に抗痙攣薬を使用することで、このリスクは低減される。
ただ、抗痙攣薬への曝露により、胎児に悪影響が生じるリスクが高まる可能性がある(薬剤の種類、服用量、服用した時点の妊娠段階により変わってくる)。
それでも、多くの場合、抗痙攣薬への曝露に起因する胎児への悪影響のリスクは、母体の発作性障害を未治療のままにしておくことによる悪影響のリスクより小さい。
したがって、一般的には、妊娠中の母体の発作性障害の治療に抗痙攣薬を使用することが推奨されている。
理想を言うと、妊娠中に抗痙攣薬を使用するリスクと利点に関する話し合いは、受胎前の段階で行っておくことが望ましい。
そうしておけば、妊娠に先立ってリスクの低い薬剤へと変更しておくといったことも可能となる[Sarmaら2016]。
頸椎奇形があると、可動域制限や不安定性につながることがあり、ひいては気道管理上の合併症を引き起こすことにもつながる可能性がある。
胸椎・腰椎領域の奇形や側彎がある場合は、脊髄くも膜下麻酔や硬膜外麻酔の際に問題になることがある。
罹患胎児は、平均妊娠週数が36週から38週のlate preterm児となるリスクを有している[Baerら2018]。
妊娠中の薬物使用に関する情報の詳細については、「MotherToBaby」を参照されたい。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「Clinical Trials.gov」、ならびにヨーロッパの「EU Clinical Trials Register」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
Wiedemann-Steiner症候群(WSS)は常染色体顕性の遺伝様式を示す。
家族構成員のリスク
発端者の両親- 両親の分子遺伝学的検査が済んでいる例について言うと、その大多数が新生の病的バリアントに起因して本疾患が生じたものである。
- 稀に、WSSと診断された罹患者の親が、やはり罹患者であったという例がみられる[Baerら2018,Sheppardら2021]。
- 仮に、家系内で発端者が唯一の罹患者(すなわち、孤発例)であるように見受けられたとしても、再発リスクに関するカウンセリングの信頼性を高める必要から、発端者の両親については分子遺伝学的検査を行っておくことが推奨される。
- 発端者の両親がどちらも見かけ上、無症候で、発端者で同定された病的バリアントがいずれの親からも検出されず、なおかつ、親子鑑定の結果、真正の生物学的父、母であることが確定しているような場合、可能性として次のようなことを頭に入れておく必要がある。
- 発端者の有する病的変異が新生のものであるという可能性
- 生殖細胞系列モザイク*を有する片親から、発端者が病的変異を継承した可能性
注:親の白血球DNAを検査したとしても、体細胞モザイクをもつ全例で病的変異が検出されるわけではない。
また、生殖細胞系列のみに病的変異を有する例については、この手法では一切検出することができない。
* 体細胞と生殖細胞系列の両方にKMT2Aの病的バリアントを有する片親については、症候の現れ方が軽度ないしごく軽微である場合がある[Baerら2018]。
- WSSと診断された罹患者の家族歴は、血族の有する軽度の症候を見逃してしまったため、一見したところ陰性であるように思われる場合がある。 そのため、両親に対して適切な臨床的評価を行うとか、分子遺伝学的検査(発端者で同定された病的変異をヘテロ接合で有している親がいないということを確認する検査)を行うといったことがない限り、家族歴陰性の確定はできない。
発端者の同胞
発端者の同胞の有するリスクは、発端者の両親の有する臨床的/遺伝的状況によって変わってくる。
- 発端者の片親が罹患者である、ないし、発端者で同定されたのと同じ病的変異を片親が有していることがわかっているといった場合であれば、同胞の有するリスクは50%である。
- 同一の病的変異を共有する同胞間にあっても、臨床症候には大きな幅がみられる。
- 発端者の有しているKMT2Aの病的バリアントが、両親いずれの白血球DNAからも検出されないといった場合であれば、同胞への再発リスクは、一般集団におけるリスクよりも若干高い程度ということになる。
- 両親に対し、ごくわずかな症候がみられないか徹底的に臨床診査を行った結果、両親とも臨床的には非罹患者と考えられたものの、分子遺伝学的検査までは行っていないといった場合であれば、発端者の同胞の有するリスクは、低いながらも一般集団の有するリスクよりは高いものと思われる。
6人の子どもがWSSという1家系で、成長障害(FTT)や摂食障害は全例でみられたものの、発達遅滞の程度には大きな幅がみられ、器質的奇形については、これをもつものともたないものがみられたという例が存在する[Sheppardら2021]。
それは、臨床的には非罹患者と目される片親が、生殖細胞系列モザイクを有している可能性が考えられるからである。
実際に、臨床的には非罹患者であるように見える片親が、KMT2Aの病的バリアントをモザイクで有していた1家系の報告が存在する[Baerら2018]。
それは、親の生殖細胞系列モザイクの可能性が考えられるからである。
発端者の子
WSSの子はそれぞれ、KMT2Aの病的バリアントを継承する50%の可能性を有する。
他の家族構成員
他の血族の有するリスクは、発端者の両親の状況によって変わってくる。
仮に片親が病的変異を有していたということになれば、その片親の血族にあたる人はすべてリスクを有するということになる。
遺伝カウンセリングに関連した問題
家族計画
- 遺伝的リスクの確定や、出生前/着床前遺伝子検査を受けるかどうかの話し合いといったことに最も適しているのは、妊娠前の時期である。
- 罹患者である若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
出生前検査ならびに着床前の遺伝子検査
家系内にあるKMT2Aの病的バリアントの内容が同定されている場合は、出生前検査や着床前遺伝子検査を行うことが可能である。
出生前検査の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。
現在、多くの医療機関では、出生前検査を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- Wiedemann-Steiner Syndrome Foundation
1314 44th Street
Sacramento 95819
Phone: 916-502-2120
Email: info@wssfoundation.org
www.wssfoundation.org - American Association on Intellectual and Developmental Disabilities (AAIDD)
Phone: 202-387-1968
Fax: 202-387-2193
www.aaidd.org - Face Equality International
United Kingdom
Email: info@faceequalityinternational.org
www.faceequalityinternational.org - MedlinePlus
Intellectual Disability - CoRDS Registry
Sanford Research
Phone: 605-312-6423
Email: sanfordresearch@sanfordhealth.org
CoRDS Registry
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:Wiedemann-Steiner症候群:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specific データベース |
HGMD | ClinVar |
|---|---|---|---|---|---|
| KMT2A | 11q23.3 | Histone-lysine N-methyltransferase 2A | KMT2A database | KMT2A | KMT2A |
データは、以下の標準資料から作成したものである。
遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。
リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B .Wiedemann-Steiner症候群のOMIMエントリー閲覧はすべてOMIMへ)
| 159555 | LYSINE-SPECIFIC METHYLTRANSFERASE 2A;KMT2A |
| 605130 | WIEDEMENN-STEINER SYNDROME;WDSTS |
分子レベルの病原
KMT2Aの遺伝子産物は、ヒストンH3のメチル化に関与するDNA結合タンパク質である。
そのため、数多くの標的遺伝子の発現を制御することになる。
KMT2Aは、Hox遺伝子の発現を制御するショウジョウバエ、トリソラックスの哺乳動物ホモログである。
KMT2Aは、造血や、軸方向の骨格のパターン化に重要な役割を果たしている[Hessら1997]。
疾患の発症メカニズム
KMT2Aの病的ナンセンスバリアントにより、ナンセンス変異依存mRNA分解機構が働く。
したがって、メカニズムとしてはハプロ不全が生じているものと考えられる[Jonesら2012]。
病的ミスセンスバリアントの場合は、それが生じる部位により変わってくる可能性があるものの、予備的研究の示すところでは、どうやら標的遺伝子に発現低下が生じている模様である。
CXXCドメインは、標的遺伝子との結合に重要な役割を果たすと考えられており、TADドメインは転写活性化の上で重要である。
CXXCドメインのミスセンスバリアント(c.3460C>T,p.Arg1154Trp)により、KMT2Aの転写産物の過剰発現、ならびに下流の標的遺伝子であるSIX2やMEIS1の発現低下が生じることがわかっており、その背景にDNAへの結合障害があると考えられている。
逆に、TADドメインのミスセンスバリアント(c.8558T>G,p.Met2853Trp)は、転写量に大きな変化は生じなかったものの、SIX2やMEIS1転写産物には明らかな減少が生じたことから、転写活性化に生じた問題であると考えられている[Lebrunら2018]。
癌ならびに良性腫瘍
小児ならびに成人の白血病の約10%は、KMT2Aの絡む90種超の融合遺伝子によって引き起こされる。
こうした融合により、エピジェネティックな制御に乱れが生じ、ひいては遺伝子の転写に異常が生じると考えられている[Winters & Bernt 2017,Meyerら2018]。
縦隔原発大細胞型B細胞性リンパ腫を有する1家系3同胞と、瀰漫性大細胞型B細胞性リンパ腫を有するそのいとこに関する1報告がみられる。
どちらも非Hodgkinリンパ腫のサブタイプで、エクソームシーケンシングにより、KMT2Aの変異(その論文の記述をそのまま引用すると、MLL 5533C>A[His1845Asn])が同定されたという。
この変異は、この家系内の3人の健康な人においても同定されている。
一方、この変異は、86人の健康なフィンランド人対照者、92人の孤発性非Hodgkinリンパ腫症例、14人の家族性非Hodgkinリンパ腫症例のスクリーニングでは見出されていない[Saarinenら2013]。
更新履歴:
- Gene Reviews著者: Sarah E Sheppard, MD, PhD and Fabiola Quintero-Rivera MD.
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2022.5.26. 日本語訳最終更新日: 2022.6.29.[ in present]
![]()