ペンドレッド症候群/非症候性前庭水管拡大症
(Pendred Syndrome/Nonsyndromic Enlarged Vestibular Aqueduct)
[Synonyms: PDS/NSEVA, PDS/DFNB4]
Gene Reviews著者: GeneReviews著者: Richard JH Smith, MD, Yoichiro Iwasa, MD, PhD, and Amanda M Schaefer, MS, LGC.
日本語訳者: 清水 日智(社会福祉法人恩賜財団済生会支部済生会長崎病院 小児科,長崎大学病院小児科)
Gene Reviews 最終更新日:2020.6.18 日本語訳最終更新日: 2020.10.4
原文: Pendred Syndrome/Nonsyndromic Enlarged Vestibular Aqueduct
要約
疾患の特徴ペンドレッド症候群 (PDS) および非症候性前庭水管拡大症 (NSEVA) は、通常は先天性でしばしば高度あるいは重度の聴力低下 (軽度から中等度で進行性の聴力低下も生じうる) を伴う様々な程度の感音難聴、前庭機能障害、側頭骨の異常 (両側前庭水管拡大に蝸牛低形成を伴うこともあれば伴わないこともある) といった症状から構成される。ペンドレッド症候群では小児期の終わりから成人期の初期にかけて甲状腺腫の発症を伴うが、非症候性前庭水管拡大症では伴うことはない。
診断・検査ペンドレッド症候群および/または非症候性前庭水管拡大症を有する発端者の少なくとも50%においては、両アレル性にSLC26A4の病原性変異が同定されるか、もしくは二重ヘテロ接合性にSLC26A4に1つの病原性変異とFOXI1またはKCNJ10に1つの病原性変異が同定されることにより、分子診断が確立されている。
ペンドレッド症候群は、発端者が、感音難聴、薄層スライスCTにて確認された特徴的な側頭骨異常、および甲状腺腫を有していた場合に臨床診断がなされる。これに対して、発端者が感音難聴と前庭水管拡大の側頭骨所見を有する場合には、非症候群性前庭水管拡大症の臨床診断となる。ペンドレッド症候群では、側頭骨の異常に前庭水管拡大と、蝸牛低形成 (蝸牛の回転数が通常の2.75回転ではなく1.5回転しかない異常) の両方が含まれうることに注意が必要である。非症候性前庭水管拡大症では、側頭骨の異常は前庭水管拡大に限定され、前庭水管の中間部が1.5mmを超える場合に前庭水管拡大と定義される。甲状腺腫は様々な程度で存在するため、甲状腺のサイズを評価するために用いられる手法および食餌中からのヨード摂取量を加味して甲状腺腫と判断される。甲状腺腫は罹患者の50%にしか認められないという研究もある。
管理
症状に応じた治療 :
聴覚訓練、補聴器の使用、聴覚障害者のために計画された教育プログラムを行う。高度から重度の難聴がある人には人工内耳手術の検討、甲状腺機能異常に対する標準的な治療を行う。
サーベイランス :
聴覚障害が進行している場合、最初は3~6ヵ月ごとに聴力検査を行い、その後は半年ごとまたは年1回の頻度で聴力検査を行う。甲状腺は初診時に超音波検査を実施し、定期的に触診および/または超音波検査を実施して容積の変化を確認する。甲状腺機能検査を2~3年ごとに実施する。
避けるべき薬剤/状況 :
頭蓋内圧の急激な上昇は聴力の急激な低下と関連している可能性があることを示唆する報告もある。このため、ウエイトリフティングや相手と接触することがあるスポーツ (コンタクトスポーツ) については、参加前に医師や医療従事者に相談すべきである。
遺伝カウンセリング
ペンドレッド症候群/非症候性前庭水管拡大症は常染色体劣性遺伝形式をとる。受胎時においては、罹患者の同胞 (兄弟姉妹) はそれぞれ25%の確率で罹患しており、50%の確率で無症候性キャリアとなり、25%の確率で罹患者でもなく、キャリアでもないとされる。家系内に固有の病原性変異が判明している場合、リスクの高い血縁者のためのキャリア検査、リスクの高い妊娠のための出生前検査、着床前遺伝子診断が可能となる。
診断
疑うべき所見
ペンドレッド症候群/非症候性前庭水管拡大症 (PDS/NSEVA) スペクトラムの診断は、以下の臨床所見、側頭骨画像所見、および内分泌所見がある場合に疑われる。
臨床所見
感音難聴は通常、先天性 (もしくは言語習得前) であり、進行性ではなく、聴性脳幹反応 (ABR: auditory brain stem response) 検査または純音聴力検査で測定される高度から重度までの難聴である。難聴の評価については、難聴と遺伝性難聴の概要を参照のこと。
側頭骨画像検査所見
側頭骨形成異常の同定と解釈には、適切な検査 (すなわち、薄層スライスCTが必要であり、日常撮像される側頭骨CTは一般的に不十分である)と、内耳の解剖に関する詳細な知識の両者が必要である。
- Mondini奇形/異形成 側頭骨の薄層スライスCTでは、Mondini奇形/異形成 (蝸牛低形成を伴う両側前庭水管拡大) が検出される。蝸牛は低形成となっており、通常予想される2.75回転ではなく1.5回転しかなく、前庭水管は拡大しており前庭水管中間部が1.5mmを超える。蝸牛低形成および前庭水管拡大の両者が存在することをMondini奇形/異形成と呼ぶ。
- 全てのペンドレッド症候群の患者において側頭骨の異常が放射線学的に検出されるが [Goldfeld et al 2005]、認められる所見には幅がある。同一のSLC26A4病原性変異をホモ接合性に有する個体を対象とした研究では、高分解能CTにより、患者の100%で蝸牛軸の欠損 (すなわち、蝸牛正中断 [mid-modiolar section] にて蝸牛の中心に位置する骨の多面体構造が明確ではない) が認められた。80%の患者では前庭水管拡大 (すなわち、前庭水管の下行部において中間部の径が1.5mm以上となる) を認めた。75%が蝸牛の前庭階の欠損 (すなわち、前庭階と中心階の間に骨性の中隔が認められない) [Goldfeld et al 2005] (図1) を有していた。
注:内耳低形成を伴う、または伴わない前庭水管拡大を、放射線画像診断したとしても、ペンドレッド症候群であると臨床診断できるわけではない。なぜならば、甲状腺の異常を伴わないこれらのタイプの側頭骨奇形を生じさせる原因は他にもあるためである (鑑別診断を参照)。
- 非症候性前庭水管拡大症は側頭骨の薄層スライスCTで検出される。前庭水管の中間部の径が1.5mmを超えると前庭水管拡大と判定する。
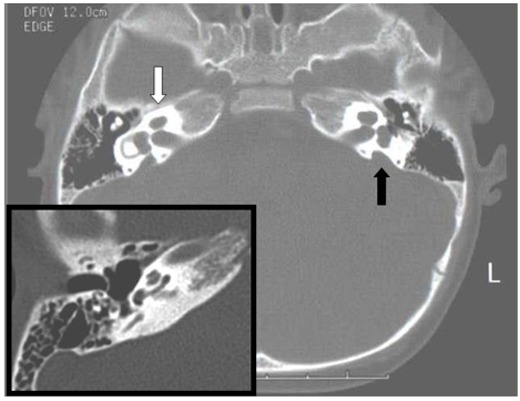
図1
ペンドレッド症候群を発症した発端者のCT (コンピュータ断層撮影) では、蝸牛の前庭階が存在せず、蝸牛軸が欠損していることがわかる (白矢印)。前庭水管拡大も認められる (黒矢印) 。差し込み図は前庭水管拡大が見られない人の正常な右の蝸牛を示しているが、この図では前庭水管拡大の有無は明らかになっていない。
内分泌学的所見
- ペンドレッド症候群における典型的な甲状腺の症状として、ヨウ素の有機化障害に起因する甲状腺機能が正常の甲状腺腫を認めることがあるが、これは甲状腺サイズを評価するための体積検査によって検出することができる。しかしながら、甲状腺腫を測定する技量は、甲状腺サイズを評価するために用いられる手法に依存する。さらに、栄養素としてのヨードの摂取は甲状腺の腫大を予防する可能性がある。いくつかの研究からは、ペンドレッド症候群患者の50%にしか甲状腺腫が発症しないことが示唆されている[Reardon et al 1999, Wémeau & Kopp 2017]。甲状腺が腫大している場合には、甲状腺ホルモン値を検査することが必要である。
- 甲状腺腫は一般的に10歳を過ぎるころから明らかになり [Suzuki et al 2007, Reardon et al 1999]、10年ごとに2.6倍に増加し続ける [Madeo et al 2009]。これらの患者の甲状腺の状態は、身体検査および超音波検査によって生涯にわたり定期的に検査され続けるべきである[Madeo et al 2009]。 (管理を参照)
注:過去においては、ヨウ素の有機化障害の診断にはパークロレイト (過塩素酸カリウム) 放出試験が用いられていた。パークロレイト試験の詳細はこちら (pdf) を参照のこと。
診断の確立
ペンドレッド症候群の臨床診断は、発端者において、感音難聴を認めること、薄層スライスCTで特徴的な側頭骨の異常を認めること、および甲状腺腫を認めることによりなされる。非症候性前庭水管拡大症の臨床診断は、感音難聴を認めること、側頭骨の所見として前庭水管拡大を認めることによりなされる (疑うべき所見を参照)。
ペンドレッド症候群/非症候性前庭水管拡大症の分子学的診断は、SLC26A4における両アレル性の病原性変異の同定、または二重ヘテロ接合性にSLC26A4に病原性変異が1つ同定され、かつFOXI1またはKCNJ10において病原性変異が1つ同定されることにより確立される (表1)。
遺伝子検査の結果は、民族集団と表現型によって異なる。
民族集団:
- 韓国人および日本人の発端者では、80%以上がSLC26A4に2つの病原性バリアントを有し、10%強が1つの病原性バリアントを有し、10%未満が病原性バリアントを有していなかった [Tsukamoto et al 2003, Park et al 2005]。
- ペンドレッド症候群/非症候性前庭水管拡大症を有する北米またはヨーロッパのコーカソイド (白人) では、常染色体劣性遺伝の場合に予想されるようなSLC26A4に2つの病原性バリアントを有するのは約25%のみである [Pryor et al 2005, Ito et al 2011]。約半数は検出可能なSLC26A4の病原性バリアントを持たず、25%では1つの病原性バリアントのみが検出される [Choi et al 2009a]。
表現型:
- コーカソイドにおける病原性変異の数は、聴覚および甲状腺の表現型と強い相関がある。すなわち、ペンドレッド症候群の患者は、非症候性前庭水管拡大症の患者よりも病原性変異を両アレル性に持つ可能性が高い [Azaiez et al 2007]。
- 非症候性前庭水管拡大症を有する人の聴覚障害の程度は、2個の (1個または0個ではなく) SCL26A4病原性変異が同定された場合により重度となる [King et al 2010, Rose et al 2017]。
これらの分子学的所見の説明は、Chattarajら [2017] によりなされている。すなわち、彼らは非症候性前庭水管拡大症を有するコーカソイド (白人) のハプロタイプを同定することにより、既知の翻訳領域の変異もしくはスプライスサイトの変異とは別のアレル上に (in trans)、非症候性前庭水管拡大症において頻繁に見られる変異ーSCLC26A4の上流に存在する12個の変異から構成されるーを同定した [King et al 2010, Rose et al 2017]。
分子遺伝学的検査へのアプローチ すべての聴覚障害がある人に対して、聴覚障害および難聴のためのマルチ遺伝子パネルを使用することは、診断費用を最小限に抑えつつ診断率を最大化する。
聴覚障害および難聴のマルチ遺伝子パネルには、通常、SLC26A4、FOXI1、KCNJ10、そしてその他の関心のある遺伝子が含まれている (鑑別診断を参照)。注: (1) この種のパネルに含まれる遺伝子の種類や診断の感度は検査室によって異なり、時代とともに変化する可能性がある。 (2) マルチ遺伝子パネルの中には、当GeneReviewのサイト上で説明した病態に関連しない遺伝子が含まれている場合がある。したがって、臨床医は、意義不明な変異が同定されてしまったり、患者の表現型を説明できない遺伝子の病原性変異が同定されてしまったりすることを防ぎながら、最も合理的な費用で病態の遺伝的原因を同定する最良の機会を提供するマルチ遺伝子パネルを決定する必要がある。 (3) パネルで使用される方法にはシーケンス解析、欠失/重複解析、および/または他のシーケンス解析に基づかない手法による検査が含まれる。
マルチ遺伝子パネルの詳細については、こちらを参照のこと。
注: 包括的なゲノムシーケンス解析 (すなわち、エクソームシーケンス解析とゲノムシーケンス解析) は、現在のところ、難聴の原因遺伝子の一次スクリーニングとしては正当化されていない[Sloan-Heggen et al 2016]。
表1.
ペンドレッド症候群 (PDS) および非症候性前庭水管拡大症 (NSEVA) で用いられる分子遺伝学的検査
| 遺伝子1,2 | 遺伝子の病原性変異に起因するPDSとNSEVAの割合 | 各手法による病原性変異の検出割合3 | ||
|---|---|---|---|---|
| PDS | NSEVA | シーケンス解析4 | 遺伝子標的欠失重複解析5 | |
| FOXI1 | 検出されていない | <1%6 | 2/26 | 不明 |
| KCNJ10 | 検出されていない | <1%7 | 2/27 | 不明 |
| SLC26A4 | ~90%8 | 50%~90%8 | ~90% | ~10%9 |
| 不明 | 不明 | ~50% | 該当なし | |
- 遺伝子名はアルファベット順に掲載されている。
- 染色体座とタンパク質については、表A.遺伝子とデータベースを参照のこと。
- この遺伝子で検出されたアレル変異の情報については、分子遺伝学的情報を参照のこと。
- シーケンス解析は、良性、良性の可能性が高い、意義不明、病原性の可能性が高い、または病原性のある変異を検出する。病原性変異には、小さな遺伝子内欠失/挿入、ミスセンス変異、ナンセンス変異、スプライスサイト変異が含まれることがあり、通常、エクソンまたは全遺伝子の欠失/重複は検出されない。シーケンス解析結果を解釈する際に考慮すべき事項については、こちらを参照のこと。
- 遺伝子を標的とした欠失/重複解析は、遺伝子内欠失または重複を検出する。定量PCR、ロングリードPCR、MLPA法 (multiplex ligation-dependent probe amplification) 、単一エクソン欠失や重複を検出するために設計された遺伝子標的マイクロアレイなどの方法がある。
- 2家系において、NSEVA患者は、SLC26A4とFOXI1の両者にヘテロ接合性の病原性バリアントを有していた [Yang et al 2007]。
- 2家系において、NSEVA患者は、KCNJ10とSLC26A4の両者にヘテロ接合性の病原性バリアントを有していた[Yang et al 2009]。
- SLC26A4遺伝子変異に起因するペンドレッド症候群/非症候性前庭水管拡大症の割合は、確認方法、遺伝、および民族集団によって異なる。内耳奇形 (特に、内耳低形成を伴うか否かに関わらず、前庭水管拡大) の有無が確認された患者のうち、SLC26A4遺伝子異常に起因する患者の割合は、欧州系アメリカ人集団では~40-50%であり、疾患の家系内集積がある集団や、アジア人集団ではより高くなる [Campbell et al 2001, Tsukamoto et al 2003, Berrettini et al 2005, Huang et al 2011, Chattaraj et al 2017, Rose et al 2017]。
- SLC26A4における単一エクソンおよび複数のエクソンの欠失が報告されている [Pera et al 2008]。
臨床的特徴
疾患の説明
ペンドレッド症候群/非症候性前庭水管拡大症 (PDS/NSEVA) は、感音難聴、前庭機能障害、側頭骨の異常といった表現型スペクトラムから構成される。また、ペンドレッド症候群では小児期の終わりから成人期の初期にかけて発症する甲状腺機能が正常の甲状腺腫の合併も含まれるが、非症候性前庭水管拡大症には含まれない。
ペンドレッド症候群 (PDS)
様々な程度の聴覚障害と甲状腺疾患が認められ、同一家系内においてもばらつきがある[Tsukamoto et al 2003, Napiontek et al 2004]。
聴覚障害 聴覚障害の程度とその症状は様々である。古典的には、聴覚障害は両側性であり高度から重度のもので、先天性 (もしくは言語習得前) に発症する。しかし、聴覚障害の発症は後天的にも生じ得るし、進行性の場合もある。幼児期に急速に進行することがあり[Stinckens et al 2001]、頭部外傷、感染症、または遅発性の二次性水頭症を伴うことがある [Luxon et al 2003]。めまいは、聴力の変動に先行したり、伴ったりすることがある [Sugiura et al 2005a, Sugiura et al 2005b]。頻繁に観察される低音域の気骨導差と、正常なティンパノメトリー検査所見との組み合わせは、前庭水管拡大によって引き起こされる「第3の窓」効果を示している可能性がある [Merchant et al 2007]。
前庭機能障害 前庭機能障害の客観的所見はペンドレッド症候群患者の66%に認められ、軽度の片側性半規管麻痺から重度の両側性前庭機能不全まで様々である。前庭機能障害は、正常な運動発達を示す乳児において歩行困難のエピソードを認めた場合には、疑わなくてはならない。
側頭骨の異常 すべてのペンドレッド症候群の患者において、放射線画像検査では側頭骨の異常を認める [Goldfeld et al 2005]。しかしながら、異常の種類について普遍的な合意は得られていない。疑うべき所見を参照のこと。
側頭骨の異常については、罹患した同胞間でも不一致がみられることがある [Goldfeld et al 2005]。
甲状腺腫 ペンドレッド症候群患者の約75%に臨床検査では甲状腺腫を認める。甲状腺腫は不完全浸透であり、約40%の患者では小児期の終わりから思春期の初期にかけて発症し、残りの患者では成人初期に発症する。
同一家系内においても甲状腺腫の発生は顕著に異なり、[Reardon et al 1999, Madeo et al 2009]、小児期に非症候性前庭水管拡大症とペンドレッド症候群を区別することは困難となっている。
ペンドレッド症候群の患者の多くは甲状腺ホルモン製剤 (サイロキシン) の投与を開始されるが、血清TSH値>5mU/Lで定義される甲状腺機能異常を伴う症例は約10%に過ぎない。
甲状腺腫が存在しない場合の甲状腺機能異常は報告されていない。
非症候性前庭水管拡大症 (NSEVA)
非症候性前庭水管拡大症は、側頭骨のCTやMRIで前庭水管拡大を認めるが、他の明らかな異常がない感音難聴が特徴である (すなわち、非症候性の聴覚障害)。甲状腺の異常は認められない。
聴覚障害 聴覚障害の程度や症状はさまざまである。非症候性前庭水管拡大症の患者の多くは、生まれつき正常な聴力を有しているが、小児期より聴覚障害が出現する。非症候性前庭水管拡大症を有する患者の大多数 (~80%) が聴力の変動があると訴えている [Rose et al 2017]。いくつかの報告では、前庭水管拡大のサイズと聴覚障害の程度との間に相関関係があると記述されているが、厳密な相関関係は確立されていない [Berrettini et al 2005]。
前庭機能障害 前庭機能障害はカロリック検査で確認されるが、前庭水管拡大を認める患者における前庭機能障害が検出されない場合がある。前庭水管拡大が片側性の場合、前庭障害がある側と、前庭の拡大がある側との間には、厳密な相関関係はない[Berrettini et al 2005]。
側頭骨の異常 前庭水管拡大は、乳児期または小児期に発症する感音難聴を有する患者に最も多くみられる一般的な画像所見である。前庭水管拡大は両側性または片側性であることがある。
遺伝型と表現型の相関性
ペンドレッド症候群/非症候性前庭水管拡大症の臨床スペクトラムにおける遺伝型と表現型の関係を理解することは、患者のケアに役立つ。
ペンドレッド症候群および非症候性前庭水管拡大症の表現型は、ペンドレッド症候群における甲状腺機能障害の合併に基づいて区別できる。甲状腺の表現型は、SLC26A4にコードされるタンパク質であるペンドリン (Pendrin) が持つ、ヨウ化物輸送能の残存量に依存する [Pryor et al 2005, Pera et al 2008]。
遺伝子変異の種類 (ミスセンス変異 vsナンセンス変異) と甲状腺腫の発症との間に強い相関性はない。そして、SLC26A4に両アレル性の病原性ないし病原性が疑われる変異を有する患者においては、変異の種類にかかわらず甲状腺に関連する症状の発症リスクが上昇する[Pryor et al 2005、Ladsous et al 2014、Suzuki et al 2007]。 (管理を参照。)
病原性変異はタンパク質を構成する780アミノ酸のいずれの場所でも起こりうる。新規の病原性を有するミスセンス変異が同定された場合、表現型 (すなわち、中等度、高度、または重度の難聴; 甲状腺肥大) の予測を、in vitroにおける追加の機能検査の実施なく行うことは、非常に困難である。
他の名称
ペンドレッド症候群 (PDS) と非症候性前庭水管拡大症 (NSEVA: nonsyndromic enlarged vestibular aqueduct) は、連続性のある疾患の一部と考えるべきである [Reardon et al 1999, Azaiez et al 2007]。
ペンドレッド症候群は「常染色体劣性感音難聴、前庭水管拡大、および甲状腺腫」とも呼ばれる。
非症候性前庭水管拡大症は、以下のようにも呼ばれる:
- 非症候性前庭水管拡大型難聴 (Nonsyndromic enlarged vestibular aqueduct hearing loss)
- 常染色体劣性遺伝性非症候性難聴4 (DFNB4: Autosomal recessive nonsyndromic deafness 4)
- DFNB4非症候性聴覚障害および前庭水管拡大
前庭水管拡大 (EVA: enlarged vestibular aqueduct) は、前庭水管拡張 (DVA: dilation of the vestibular aqueduct) とも呼ばれる。
有病率
ペンドレッド症候群/非症候性前庭水管拡大症を同じ疾患スペクトラムの一部と考えた場合、SLC26A4の病原性変異が難聴の3番目に多い原因となっているため、有病率は非常に高くなる (図2)。
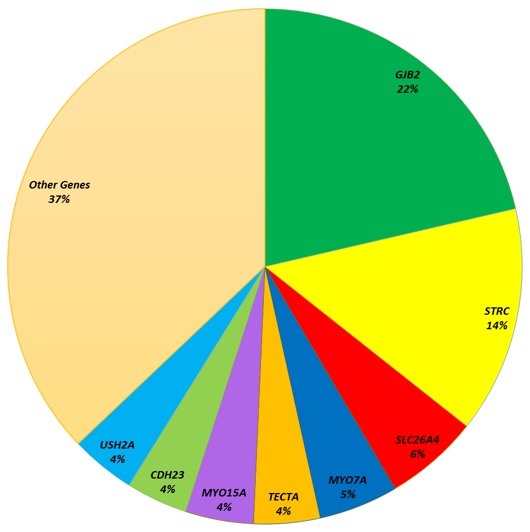
図2. 難聴に対する包括的な遺伝学的検査を受けた2434人の無作為抽出のスクリーニング検査において、SLC26A4の両アレル性病原性変異に起因するペンドレッド症候群/非症候性前庭水管拡大症は、79種類の鑑別となる難聴の原因遺伝子の中で3番目に多く、全体の6%を占めていた[Sloan-Heggen et al 2016; Smith et al, unpublished data]。
遺伝子に関連する (対立遺伝子) 疾患
FOXI1 FOXI1の病原性変異は、ペンドレッド症候群/非症候性前庭水管拡大症に関連してのみ同定されている。 (二遺伝子性:digenic [2つの異なる遺伝子における病原性変異が表現型の発現に必要となる] の形態をとる)
KCNJ10 KCNJ10における両アレル性病原性変異は、SeSAME症候群 (けいれん発作[seizures]、感音難聴 [sensorineural deafness]、運動失調 [ataxia]、精神発達遅滞 [mental retardation]、電解質異常[electrolyte imbalance]) [Scholl et al 2009]およびEAST症候群 (強直間代けいれん [epilepsy]・小脳失調 [ataxia]・感音難聴 [sensorineural deafness]・尿細管障害 [tubulopathy]) [Freudenthal et al 2011](OMIM 612780)の原因となっている。
SLC26A4 SLC26A4の病原性変異に関連することが知られている唯一の表現型は、ペンドレッド症候群/非症候性前庭水管拡大症スペクトラムである。
鑑別診断
先天性遺伝性聴覚障害 先天性 (もしくは言語習得前) 遺伝性聴覚障害は、新生児の約1,000人に1人が罹患している。これらの新生児の30%にはさらに異常があり、症候性難聴の診断が可能となる(「難聴と遺伝性聴覚障害の概説」を参照)。
前庭帯水管拡大は、蝸牛低形成を伴うものと伴わないものがあり、実質的にすべてのペンドレッド症候群患者に見られるが、前庭水管拡大も蝸牛低形成もペンドレッド症候群に特異的なものではない。これらのタイプの側頭骨奇形を引き起こす他の原因としては、先天性サイトメガロウイルス感染症や、甲状腺異常を伴わない先天性鰓弓耳腎 (Branchio-oto-renal: BOR) 症候群などがある。
感音難聴を伴う先天性甲状腺機能低下症 感音難聴を伴う散発性や風土性の先天性甲状腺機能低下症は、臨床的にはペンドレッド症候群に類似するが、遺伝的には異なる。
甲状腺ホルモン不応症 甲状腺ホルモン不応症 (RTH: resistance to thyroid hormone) は、一般的に常染色体優性遺伝形式を示すが、RTHが常染色体劣性遺伝形式を示した近親婚の家系が例外的に1例報告されてい。6名の子のうち2名は重度の感音難聴と甲状腺腫を有し、甲状腺ホルモン受容体β遺伝子 (THRB; OMIM 190160) を含む3番染色体上の大きな欠失 (核型解析にて検出された) を有していた。
自己免疫性甲状腺疾患 バセドウ病、橋本病、および萎縮性甲状腺炎 (原発性特発性粘液水腫) を含む自己免疫性甲状腺疾患は、複数の遺伝的要因および環境的要因によって引き起こされる。この疾患群に関与する遺伝子の候補には、免疫応答および/または甲状腺生理を調節する遺伝子が含まれる。
これらの表現型に関連する遺伝子についてOMIM上で確認する場合は、OMIMのPhenotypic Seriesより「常染色体劣性遺伝形式をとる難聴」を参照のこと。
管理
初期診断後の評価
分子学的に診断が確立されたペンドレッド症候群/非症候性前庭水管拡大症 (PDS/NSEVA) または臨床的に診断されたペンドレッド症候群を有する患者の合併症の程度を確認するために、まだなされていない場合には、以下の評価を行うことが推奨される:
- 聴覚の評価(聴性脳幹反応検査、耳音響放射検査、純音聴力検査)
- 甲状腺のサイズを測定する甲状腺超音波検査と甲状腺機能検査 (T3、T4、TSH)
- 必要に応じて内分泌専門医へコンサルトを行う
- 臨床遺伝専門医および/または遺伝カウンセラーへコンサルトを行う
合併症の治療
以下を行うことは適切である:
- 聴覚リハビリテーション (できるだけ早い段階での補聴器の使用)
- 高度~重度難聴者における人工内耳埋込術の検討
- 聴覚障害者のための教育プログラム
- 甲状腺腫および/または甲状腺機能異常について、内科的および/または外科的治療 (内分泌専門医へのコンサルトが必要)
サーベイランス
サーベイランスには以下のようなものがある:
- 聴覚と甲状腺機能の一生涯に渡るモニタリング
- 遺伝難聴に詳しい医師による年1回の検査
- 初期のころは3~6ヶ月ごとに、その後は毎年1回繰り返される聴力検査
- ペンドレッド症候群に精通した内分泌専門医による1~2年に1回の検査
- 甲状腺容積をモニタリングするために行われる、身体検査および/または超音波検査による甲状腺サイズの評価
- 甲状腺機能検査 (T3、T4、TSH) を2~3年ごとに行う [Choi et al 2011b]
避けるべき薬剤/状況
前庭水管拡大 (EVA) を有する人では、頭蓋内圧の上昇が聴力低下の引き金になることがあるという裏付けに乏しい報告に基づいて、ウエイトリフティングや相手と接触することがあるスポーツ (コンタクトスポーツ) を避けることを推奨する医師もいる。この推奨の効果については議論の余地があり、個人差も考慮する必要があるだろう。
リスクのある血縁者の評価
リスクのある血縁者は、聴覚障害、前庭機能障害、および甲状腺異常について、初診時に罹患者と同様の方法で評価すべきである (初期診断後の評価を参照)。
家系内における病原性変異が判明している場合には、生後まもなく同胞 (兄弟姉妹) の分子遺伝学的検査が行われることにより、こどもと家族にとって適切で早期の支援と管理が提供できることとなる。
遺伝子カウンセリングを目的としたリスクのある血縁者の検査に関連する問題については、遺伝カウンセリングを参照のこと。
今後の導入が検討されている治療法
米国のClinicalTrials.govと欧州のClinical Trials Registerを検索することで、幅広い疾患や症状の臨床試験に関する情報にアクセス可能である。注:この疾患に関する臨床試験が存在しない可能性もある。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
ペンドレッド症候群/非症候性前庭水管拡大症 (PDS/NSEVA) は、一般的に常染色体劣性遺伝形式をとる。
SLC26A4 の両アレル性病原性変異は約50%の罹患者に認められ、SLC26A4 と FOXI1 または KCNJ10 の二重ヘテロ接合性変異は 1%未満の罹患者に認められる。
両アレル性にSLC26A4 病原性変異を持つ発端者の家族へのリスク
発端者の両親
- 両アレル性にSLC26A4の病原性変異を持つこどもの両親は、ヘテロ接合性に変異を有している (すなわち、SLC26A4に1つの病原性変異を持つキャリアである)。
- ヘテロ接合性に変異を持つ場合は無症状である。
発端者の同胞
- 受胎の時点において同胞はそれぞれ、ペンドレッド症候群/非症候性前庭水管拡大症を有している確率が25%、無症候性キャリアである確率が50%、罹患しておらずキャリアでもない確率が25%である。
- ヘテロ接合性に変異を持つ場合は無症状である。
発端者の子
両アレル性にSLC26A4の病原性変異を持つ患者の子は、SLC26A4 病原性変異をヘテロ接合性に持つこととなる (キャリア)。
発端者の他の家族
発端者の両親の同胞はそれぞれ、SLC26A4病原性変異のキャリアである確率が50%である。
二遺伝子性遺伝形式 (Digenic inheritance) をとる場合の血縁者のリスク
発端者の両親
片方の親はSLC26A4病原性変異のヘテロ接合体であり、もう片方の親はFOXI1またはKCNJ10病原性バリアントのヘテロ接合体であるか、もしくは片方の親が2つの病原性変異を有している可能性がある。両親ともに確認のための遺伝学的検査を受けるべきである。
発端者の同胞
各親が1つの病原性変異を有していると仮定した場合、受胎時点において同胞がそれぞれペンドレッド症候群/非症候性前庭水管拡大症を有している確率は25%、無症候性キャリアである確率は50%、罹患しておらずキャリアでもない確率は25%となる。
発端者の子
子が病原性変異を1つ受け継ぐリスクは 50%、病原性変異を2つ受け継ぐリスクは25%となる。
発端者の他の家族
発端者の両親の同胞はそれぞれ、発端者の親の遺伝的状態に応じて、0、1、または2種類の病原性変異を持つこととなる。
キャリアの同定
ペンドレッド症候群/非症候性前庭水管拡大症を有する患者の親族にして、キャリア検査を行う場合、家系内のSLC26A4、FOXI1、またはKCNJ10の病原性変異を事前に同定する必要がある。
病原性変異が同定されている個人がこどもを持つことを考えている場合、パートナーに対してキャリア検査を行うことが可能な場合がある。
遺伝カウンセリングに関連した問題
早期診断と治療を目的としたリスクのある血縁者の評価については、管理、リスクのある血縁者の評価を参照のこと。
以下の点に注意すべきである:
- ろう者の社会に属する人々や、手話を必要とする人々とのコミュニケーションには、熟練した通訳が必要である。
- ろう者の社会に属する人々は、聴覚障害を「治療」や「療養」、「予防」を必要とするハンディキャップや障害、病状ではなく、個性のひとつとして捉えている。
- 多くのろう者は、予防や生殖、家族計画についての情報よりも、医療、教育、社会サービスなどの情報を含め、自分自身のろうの原因についての情報を得たいと考えている。
- 特定の用語を使うことが好ましい。例えば、リスク (risk) に対して確率 (probability) や機会 (chance) 、聴覚障害 (hearing impaired) に対してろう者 (deaf) や難聴 (hard-of-hearing) などである。「異常 (abnormal)」などの用語は避けるべきである。
家族計画
- キャリアの状態を明確にし、出生前検査の利用可能性について議論する最適な時期は、妊娠前である。
- 聴覚障害があったり、キャリアの可能性があったりする若年成人に遺伝カウンセリングを行うことは適切である。
DNAバンキング
DNAバンクとは、将来使用する可能性を考慮し、DNA (通常は白血球から抽出されたもの) を保管することである。検査の手法や、遺伝子に対する理解、アレル変異に対する理解、疾患についての理解は将来的に向上すると思われるため、患者由来DNAをバンク化することを検討するべきである。
出生前診断および着床前診断
ペンドレッド症候群/非症候性前庭水管拡大症を有する家系の中で病原性変異の両者が同定されている場合、リスクの高い妊娠のための出生前検査と着床前遺伝子検査が可能となる。
医療の専門家の間や家族内においても、特に検査が早期診断ではなく妊娠中絶を目的とした場合には、出生前検査に対する考え方の相違が存在しうる。多くの専門機関は出生前診断については夫婦の自己決定の問題だと考えているが、この問題については議論することが適切である。
支援団体
GeneReviews のスタッフは、本疾患を持つ患者とその家族のために、疾患特異的または包括的な支援を行う組織やレジストリーを、以下の通り抽出した。他の組織が提供する情報について、GeneReviewsが責任を負うものではない。選定基準についてはこちらを参照のこと。
- My46 Trait Profile
- National Library of Medicine Genetics Home Reference
- NCBI Genes and Disease
- American Society for Deaf Children (ASDC)
800 Florida Avenue NortheastSuite 2047
Washington DC 20002-3695
Phone: 800-942-2732 (Toll-free Parent Hotline); 866-895-4206 (toll free voice/TTY)
Fax: 410-795-0965
Email: info@deafchildren.org; asdc@deafchildren.org
www.deafchildren.org
- National Association of the Deaf (NAD)
8630 Fenton Street
Suite 820
Silver Spring MD 20910
Phone: 301-587-1788; 301-587-1789 (TTY)
Fax: 301-587-1791
Email: nad.info@nad.org
www.nad.org
分子遺伝学的情報
Molecular GeneticsおよびOMIMの表の情報は、GeneReviewの表の情報とは異なる場合がある。すなわち、GeneReviewの表にはより新しい情報が含まれている場合がある。-編集者。
表A. ペンドレッド症候群/非症候性前庭水管拡大症: 遺伝子とデータベース
| 遺伝子名 | 染色体座 | タンパク質 | 遺伝子座特異的データベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| FOXI1 | 5q35.1 | フォークヘッド型転写因子I1 (Forkhead box protein I1) |
FOXI1 database | FOXI1 | FOXI1 |
| KCNJ10 | 1q23.2 | ATP感受性内向き整流性カリウムチャネル10 (ATP-sensitive inward rectifier potassium channel 10) |
KCNJ10 database | KCNJ10 | KCNJ10 |
| SLC26A4 | 7q22.3 | ペンドリン (Pendrin) |
Hereditary Hearing Loss Homepage (SLC26A4) CCHMC - Human Genetics Mutation Database (SLC26A4) Deafness Variation Database (SLC26A4) |
SLC26A4 | SLC26A4 |
データは以下の標準的な参照サイトより収載した:遺伝子はHGNCから、染色体遺伝子座はOMIMから、タンパク質はUniProtから。リンク先のデータベース(Locus Specific, HGMD, ClinVar)の説明はこちらを参照のこと。
表B. ペンドレッド症候群/非症候性前庭水管拡大症に関連するOMIM項目 (OMIMですべてを見る)
| 274600 | ペンドレッド症候群; PENDRED SYNDROME; PDS |
| 600791 | 難聴、常染色体劣性4,前庭水管拡大を伴う; DEAFNESS, AUTOSOMAL RECESSIVE 4, WITH ENLARGED VESTIBULAR AQUEDUCT; DFNB4 |
| 601093 | フォークヘッド型転写因子I1; FORKHEAD BOX I1; FOXI1 |
| 602208 | カリウムチャネル、内向き整流性、サブファミリーJ、ナンバー10; POTASSIUM CHANNEL, INWARDLY RECTIFYING, SUBFAMILY J, MEMBER 10; KCNJ10 |
| 605646 | 溶質輸送体ファミリー26; SOLUTE CARRIER FAMILY 26, MEMBER 4; SLC26A4 |
分子病態
蝸牛内電位 (endocochlear potential) の喪失が、ペンドレッド症候群/非症候性前庭水管拡大症 (PDS/NSEVA) における難聴の原因である [Wangemann et al 2004]。内リンパK+濃度が正常であることは、ペンドリンの欠失または機能不全が、二次的にKCNJ10タンパク質の発現を低下させ蝸牛内電位の障害をもたらすことを示唆している。
甲状腺の表現型 (PDSおよびNSEVA) は、SLC26A4によってコードされるタンパク質であるペンドリンのもつヨウ化物輸送能の残存量に依存する [Pryor et al 2005, Pera et al 2008]。SLC26A4の両アレル性機能喪失型変異は、ヨウ素の有機化障害および甲状腺濾胞へのヨウ化物運送の減少による甲状腺機能障害と常に関連している。ペンドリンが存在しない場合、塩素チャネル5 (ClC-5) の発現が増加し、一過性に甲状腺濾胞内腔側へのヨウ化物の運送 (apical iodide efflux) を代償することとなる。より深刻な影響を受けた甲状腺濾胞では、デュアルオキシダーゼ (Duox) と甲状腺ペルオキシダーゼ (TPO) が細胞質内に再配置され、異常な細胞内甲状腺ホルモン合成を引き起こし、細胞破壊をもたらす [Senou et al 2010]。
しかしながら、変異の種類(ミスセンス変異 vs ナンセンス変異)と甲状腺腫の発症との間の相関は強くはなく、SLC26A4の病原性/病原性の可能性のある変異型を両アレル性に有している患者は、変異の種類に関係なく甲状腺関連の症状を発症するリスクが高い。
PDS/NSEVAの患者において、SLC26A4とKCNJ10の両方で単一の病原性変異を有する二重ヘテロ接合性が確認されたことは、PDS/NSEVAの病因においてKCNJ10の関与を証明する追加的な証拠となった [Yang et al 2009]。Yangら[2007]はまた、SLC26A4だけではなく、その転写制御機構を調節するFOXI1も関与するPDS/NSEVAの存在を明らかにしており、SLC26A4タンパク量依存的に発症するというPDS/NSEVAの分子病態モデルと一致している。
二遺伝子性遺伝 (Digenic inheritance) 罹患した患者が二重ヘテロ接合性 (関連する遺伝子のうちの2つの遺伝子がそれぞれヘテロ接合性となる) である、明らかな二遺伝子性の遺伝形式を認めたまれな報告が存在する。
- 2家系において、前庭水管拡大 (EVA) を有する患者において、SLC26A4の病原性変異とFOXI1の病原性変異が二重ヘテロ接合性に同定された。この遺伝型の病原性の裏付けとして、研究者らは、FOXI1が5'側に存在する保存されたプロモーター作用を持つシスエレメントに結合することでSLC26A4の転写が活性化されることを示した [Yang et al 2007]。Yangら[2007] は、PDS/NSEVAを有する他の家系において、FOXI1により引き起こされる転写活性化を無効にする、SLC26A4のプロモーターサイトにおける病原性変異を同定した。
- 他の2家系において患者は、SLC26A4の病原性変異とKCNJ10の病原性変異を二重ヘテロ接合性に有していた。同定されたSLC26A4の病原性変異は、過去にPDS/NSEVAに関与していることが示唆されていた。KCNJ10の病原性変異は、蝸牛内電位 (endocochlear potential) の発生と維持に重要なK(+) コンダクタンス活性を低下させる [Yang et al 2009]。
FOXI1またはKCNJ10の病原性変異は、前庭水管拡大の稀な原因となる [Jonard et al 2010, Wu et al 2010]。
FOXI1
遺伝子の構造 フォークヘッドボックスL1遺伝子であるFOXI1は、1038ヌクレオチドで構成される翻訳領域を持つ1つのエクソンからなる (NM_005250.2)。2つのトランスクリプトバリアントは、異なるアイソフォームをコードしている (アイソフォーム "a "は378アミノ酸、アイソフォーム "b "は283アミノ酸からなる)。遺伝子およびタンパク質情報の詳細な要約については、表A 遺伝子を参照のこと。
病原性変異 これまでのところ、FOXI1における両アレル性病原性変異は同定されていない。しかしながら、5つの非同義変異 (non-synonymous variants) と推定される変異が同定されており、そのうちの1つはフォークヘッドDNA結合ドメインにおける1アミノ酸欠失であった [Yang et al 2007]。FOXI1が結合するSLC26A4プロモーターの転写調節エレメントは、2つの隣接するFOXI1結合部位が頭を向かい合わせるように配置されている。この結合部位はFBS1およびFBS2と呼ばれ、この結合部位の両者が、この特別な向きで位置していることが、FOXI1が介在するSLC26A4の転写活性化に必要である。SLC26A4の翻訳領域における病原性変異および他方のアレルのFBS1またはFBS2の病原性変異が、PDS/NSEVAのまれな原因となる [Yang et al 2007] (分子病態、二遺伝子性の遺伝を参照のこと)。
正常遺伝子産物 345アミノ酸からなるタンパク質、FOXI1 (NP_005241.1) をコードするFOXI1は、蝸牛と前庭の発生に必要な初期の耳胞特異的遺伝子であり、SLC26A4の上流調節因子である。FOXI1タンパク質の機能の一つは、SLC26A4の発現を制御する転写因子としての機能である [Yang et al 2007]。
異常遺伝子産物 in vitroの研究では、病原性があると推定された5つの変異のうちの1つの遺伝子配列をもつFOXI1転写産物より翻訳されたFOXI1アイソフォームは、FOXI1トランス活性作用を介在するSLC26A4の発現を損なうことが実証された。これらのデータは、FOXI1の欠損とPDS/NSEVAとの因果関係を支持するものである[Yang et al 2007]。選択的Foxi1欠損ホモ接合性マウスは、感音難聴と前庭上水管の拡大を呈する [Hulander et al 2003]。
KCNJ10
遺伝子の構造 KCNJ10は2つのエクソンで構成されており(NM_002241.4) 、そのうちの最初のエクソンは非翻訳 (non-coding) である。遺伝子およびタンパク質情報の詳細な要約については、表A 遺伝子を参照のこと。
病原性変異 これまでのところ、KCNJ10の両アレル性病原性変異は、PDS/NSEVAを有する人においては同定されていない。しかしながら、1つのKCNJ10病原性変異および1つのSLC26A4病原性変異を二遺伝子性に受け継いだことによりPDS/NSEVAが発症したとする2例の報告がある (分子病態を参照のこと) [Yang et al 2009]。
KCNJ10の病原性変異によって生じる他の表現型については、遺伝的に関連する疾患を参照のこと。
正常遺伝子産物 KCNJ10は379アミノ酸からなるタンパク質 (NP_002232.2) をコードしており、このタンパク質は内向き整流性カリウムチャネルファミリーのひとつであり、カリウムの細胞内への (細胞外にではなく) 流入を増加させる特徴を持つ(RefSeqより; 7/2008)。蝸牛の血管条、腎臓の遠位曲尿細管、脳のグリア細胞で発現している。
異常遺伝子産物 in vitroの機能アッセイにより、PDS/NSEVAを有する家族において同定された2つのKCNJ10病原性変異は、チャネル活性に有害であり、K+コンダクタンスを約50%低下させるタンパク質をコードしていることが証明されている [Yang et al 2009]。
SLC26A4
遺伝子の構造 SLC26A4は21個のエクソンからなる。エクソン1は非翻訳 (non-coding)であり、マイナス鎖上に位置しているSLC26A4-AS1 (SLC26A4アンチセンスRNA1) と部分的に重なる。mRNA産物は約5kbの長さで、2343塩基のオープンリーディングフレームを持ち、780アミノ酸からなるペンドリンタンパク質を産生する。遺伝子とタンパク質の詳細な情報については、表A 遺伝子を参照のこと。
病原性変異 PDS/NSEVAに関連する何百もの病原性変異がSLC26A4遺伝子上に報告されている(Deafness Variation Database参照)。 3つの病原性変異、p.Leu236Pro (26%)、p.Thr416Pro (15%)、およびc.1001+1G>A (14%)は、北欧系の人において他の病原性変異よりも多く見られ、この集団においては、分子学的に確定診断がなされたPDS/NSEVA患者の中で、原因となるアレル変異の50%を占めている [Coyle et al 1998, Campbell et al 2001]。これらの遺伝子変異の再発率はそれぞれ異なるが、共通のハプロタイプ上に発生しており、これらの独立した特定の家系における、共通の創始者の存在が示唆される [Coyle et al 1998, Van Hauwe et al 1998, Park et al 2003]。
他の集団でもまた、いくつかの多く認められる創始者変異を反映したユニークで多様な病原性変異が存在する。
- c.919-2A>G、p.His723Arg、およびp.Val239Aspは、中国人、日本人/韓国人、およびパキスタン人集団の間で多く認められる病原性変異である [Tsukamoto et al 2003、Park et al 2005、Wu et al 2005、Anwar et al 2009、Choi et al 2009b]。
- p.Glu384Glyは北欧人に多く見られる [Coyle et al 1998]。
- p.Gln514Lysはスペイン人に多い [Pera et al 2008]。
SLC26A4では単一および多数のエクソン欠失が報告されている [Pera et al 2008]。
めまいのエピソードの頻度や、聴覚障害の進行程度は、病原性変異に依存している可能性がある [Sugiura et al 2005b]。
SLC26A4の病原性変異が1つしか同定されていないPDS/NSEVA罹患者が複数名存在する家系において、前庭水管拡大の表現型と、SLC26A4に関連した縦列型反復配列 (short tandem repeat) マーカーに基づいたハプロタイプの偏在が合致していたことは、調節領域における病原性変異の存在を示唆している [Choi et al 2009a]。
SLC26A4の上流にある12個のバリアントからなるコーカソイドの前庭水管拡大 (CEVA: Caucasian enlarged vestibular aqueduct) ハプロタイプは、他方のアレル上に翻訳領域 (coding) またはスプライス部位の病原性変異を持つNSEVA患者において頻繁に見られる。CEVAハプロタイプをヘテロ接合性に有する頻度は白人コントロールで5%を超えており、このハプロタイプは遺伝性難聴に関与する遺伝子の中で最も多い病原性対立遺伝子である可能性がある [Chattaraj et al 2017]。このハプロタイプは東アジアの集団 (例えば、韓国人や日本人) では観察されていないため、SLC26A4の両アレル性変異が同定される頻度が、民族集団によって異なっていることのおそらく説明となるだろう。
臨床上の転帰に影響を及ぼすSLC26A4に関連しないハプロタイプの根拠については、FOXI1, 病原性変異を参照のこと。
表2. このGeneReviewにて議論されているSLC26A4の病原性変異一覧
| DNA ヌクレオチド変異 (Alias1) |
予想されるタンパク質の変化 | 参考配列 |
|---|---|---|
| c.707T>C | p.Leu236Pro | NM_000441.1 NP_000432.1 |
| c.716T>A | p.Val239Asp | |
| c.919-2A>G | ||
| c.1001+1G>A (IVS8+1G>A) |
||
| c.1151A>G | p.Glu384Gly | |
| c.1246A>C | p.Thr416Pro | |
| c.1540C>A | p.Gln514Lys | |
| c.2168A>G | p.His723Arg |
表に記載されている変異は、著者によって提供されたものである。GeneReviewsのスタッフは、変異の分類を独自には検証していない。
GeneReviewsは、Human Genome Variation Society (varnomen.hgvs.org) の標準的な命名規則に従っている。命名法についての説明はQuick Referenceを参照のこと。
(1).現在の命名法に適合していない変異の名称
正常遺伝子産物 SLC26A4は、塩化物、ヨウ化物、重炭酸塩、ギ酸塩のトランスポーターとして機能する780アミノ酸 (86 kDa) 膜貫通タンパク質であるペンドリン (Pendrin) をコードしている。
SLC26A4は、溶質輸送体ファミリー26 (SLC26: solute carrier 26 gene family)に属し、他の13のSLC26タンパク質と有意な相同性を有する。ヒトSLC26ファミリーメンバーは、Cl-/HCO3-交換、I-/HCO3-交換、およびSO42-/HCO3-交換を含む一連の主要な陰イオン輸送活性に関与しており、PDS/NSEVA、軟骨異形成症、および先天性クロール下痢症 (congenital chloride diarrhea) を含む消耗性の疾患と関連している [Dawson & Markovich 2005, Kere 2006]。
異常遺伝子産物 SLC26A4における病原性変異は、ペンドリンの部分的または完全な活性喪失をもたらす可能性がある。機能の研究により、ヨウ化物輸送能の残存を保持するSLC26A4ミスセンス病原性変異は、PDSよりもPDS/NSEVAと関連している可能性が高いことが示唆されている [Scott et al 2000]。すべての短縮型変異は、ペンドリンの機能を喪失する。
マウスモデルを用いた研究から、胚発生16.5日目から生後2日目までの間にペンドリンが存在している事が正常な聴力獲得には必要であるが、聴力の維持には必要ではないことが示されている [Choi et al 2011a]。ホモ接合性のSlc26a4欠損マウス(null mice; Slc26a4-/-)の内リンパ量は増加し、I型およびII型線維芽細胞が占める領域の組織量は減少する。Slc26a4-/-マウスは、通常カリウムチャネルKCNJ10によって蝸牛血管条の基底細胞を越えて生成され、血管条の中間細胞に局在する、蝸牛内電位 (endocochlear potential) を欠いている。
更新履歴
- Gene Review著者: Richard JH Smith, MD, Guy Van Camp, PhD
日本語訳者: 佐藤亜位,山崎雅則(信州大学医学部附属病院加齢総合診療科),櫻井晃洋(信州大学医学部附属病院遺伝子診療部) )
Gene Review 最終更新日: 2009.4.2. 日本語訳最終更新日: 2009.10.20. -
Gene Reviews著者: GeneReviews著者: Richard JH Smith, MD, Yoichiro Iwasa, MD, PhD, and Amanda M Schaefer, MS, LGC.
日本語訳者: 清水 日智(社会福祉法人恩賜財団済生会支部済生会長崎病院 小児科,長崎大学病院小児科)
Gene Reviews 最終更新日:2020.6.18 日本語訳最終更新日: 2020.10.4(in present)