ハッチンソン-ギルフォード プロジェリア症候群
(Hutchinson―Gilford Progeria syndrome)
Gene Reviews著者: Leslie B Gordon, MD, PhD, W Ted Brown, MD, PhD, and Francis S Collins, MD, PhD.
日本語訳者:小崎 里華(国立成育医療研究センター 遺伝診療科)
GeneReviews最終更新日: 2023.10.19. 日本語訳最終更新日:2024.10.3.
原文: Hutchinson―Gilford Progeria syndrome
要約
疾患の特徴
ハッチンソン-ギルフォード早老症候群(HGPS)は、通常、小児期に発症し、早老症状に似た臨床所見を特徴とする。HGPSの小児は通常、出生時は正常である。生後1年以内に重度の成長障害を呈する。特徴的な顔貌としては、顔に対して相対的大頭、狭い鼻隆起、狭い鼻尖、薄い上下口唇、小さな口、および下顎の後退、小顎症である。一般的な特徴には、皮下脂肪の減少、歯牙の萌出遅延と乳歯の喪失、腹部と大腿上部に小さな膨らみを生じる異常な皮膚、禿頭、爪の形成異常、股関節外反、および進行性の関節拘縮を含む。その後、低周波の伝音性難聴、歯叢生および永久歯の一部欠損を呈する。精神運動発達は正常である。重度のアテローム性動脈硬化症の合併症としての心臓病(心筋梗塞、心不全)または脳血管疾患(脳卒中)によって、ロナファルニブ未治療または心臓介入手術なしでは、一般に6歳から20歳の間(平均約14.5歳)に死亡する。ロナファルニブ治療により平均寿命は17-19.5歳まで延長された。
診断・検査
古典的又は非古典的HGPSの遺伝子型の診断は、特徴的な臨床症状を有する発端者に異常ラミンAタンパク質であるプロジェリンを産生するヘテロ接合性の病的ヴァリアントを同定することによる。
古典的な遺伝子型HGPSは、ヘテロ接合性のLMNAの病的ヴァリアントc.1824C>Tを有する(HGPSの約90%)。非古典的遺伝子型HGPSは、HGPSの特徴的な臨床的症状をもち、LMNAのエクソン11またはイントロン11にあるプロジェリンを産生するヘテロ接合性の異なる病的ヴァリアントを有する(HGPSの約10%)。
臨床的マネジメント
症状に対する治療:
標的治療としてHGPSではロナファルニブが承認されている。低音の聴覚改善、頭痛の減少、頸動脈-大腿動脈間脈波伝播速度の減少、動脈壁硬度の減少、寿命の延長が認められている。
支持療法として、定期的に少量の食事を頻回に摂取する食事療法が推奨される。歯叢生を避けるため、永久歯萌出後に乳歯の抜歯を勧める。屋外での活動には、頭部を含む皮膚のすべての露出部分に日焼け止めの使用をすすめる。股関節脱臼には、理学療法と体幹装具が最も勧められる。股関節再建手術は可能だが、高リスク集団における手術の合併症を考慮する必要がある。体脂肪の減少により、下肢の疼痛を生じるかもしれない。靴パッドの使用により、この疼痛は軽減できる。日常的な理学療法と作業療法、積極的なストレッチおよび水中運動による強化エクササイズが推奨される。
脳卒中のリスクを最小限に抑えるために、身体活動を奨励しながら、最適な水分補給を維持する。心臓血管および神経血管の合併症に抗凝固療法は必要である。投薬量は年齢ではなく、体重または体表面積に基づく。ニトログリセリンは狭心症に有用で、うっ血性心不全の治療には、通常うっ血防止療法が行われる。全身麻酔と挿管は、細心の注意を払い、可能であれば、気管支ファイバー挿管が理想的である。ドライアイは、眼の潤滑剤で治療する。補聴器は、臨床的に必要な場合に使用する。通常、身体的に適応した年齢に応じた学校教育が推奨される。
二次合併症の予防:
心血管および脳卒中の合併症の予防には、低用量のアスピリン(2〜3 mg / kg体重)が推奨される。硬化した末梢血管系は脱水症に対する耐性が低い可能性があり、経口で最適な水分補給を維持することを勧める。
サーベイランス:
半年ごとの心電図、年ごとの心エコー、頸動脈二重超音波検査、神経学的検査、頭頸部MRI / MRA、脂質プロファイル、歯科検査、無血管性壊死および進行性股関節外反を評価するための股関節X線、骨密度測定の二重X線吸収測定法/末梢皮膚CT、関節拘縮の理学療法評価、眼科検査、聴力検査、および日常生活の活動の評価。
避けるべきエージェント/状況:
脱水症; 背の高い/大きな仲間が集う大勢の集団による怪我のリスク、股関節脱臼のリスクを伴うトランポリンや乗馬。身体活動は自己で制限管理するべきである。
遺伝カウンセリング
ほぼすべてのHGPSは、新規の常染色体優性遺伝の病的のヴァリアントによる。発端者の同胞への再発リスクは小さいが(HGPSは通常、新規の病的ヴァリアントによって生じるため)、親の生殖細胞系列モザイクの可能性があるため、一般集団のリスクよりも高い。LMNAの病的のヴァリアントが家系内に同定されている場合、一般集団より高いリスクのため、妊娠中の出生前検査は可能である。
Gene reviewスコープ
ハッチンソン-ギルフォード プロジェリア症候群:含まれる遺伝子型
- ハッチンソン-ギルフォード プロジェリア症候群(HGPS)、古典的
- 非典型ハッチンソン-ギルフォード プロジェリア症候群
同義語と古い名前については、命名法を参照。
診断
臨床診断
示唆する所見
ハッチンソン-ギルフォードプロジェリア症候群(HGPS)は、重度の成長不全、硬化性皮膚所見、2歳までに全頭部の禿頭に進行する部分的脱毛症、全身性脂肪ジストロフィー、下顎後退、X線所見で外反股、遠位端の骨溶解を伴う短い鎖骨および乳歯の遅延/不完全な萌出、正常な知的発達。
成長不全
- 低身長(<3パーセンタイル)
- 体重増加不良(<3パーセンタイル)、身長に対して顕著な低体重
- 全身的な皮下体脂肪の減少
顔の特徴(図1を参照)
- 顔に対して不均衡な大頭
- 細い鼻
- 薄い上下口唇
- 下顎後退と小顎
外胚葉
- 歯科 乳歯の萌出遅延と乳歯の脱落遅延、永久歯の一部萌出遅延、歯叢生
- 皮膚 さまざまな色素沈着、張り詰めた皮膚、硬化した皮膚、下腹部および/または大腿近位部の皮膚の小さな膨らみ
- 毛髪 全禿頭、時々、非常に粗な産毛のような未熟な髪が残っている。眉毛の喪失、爪の形成異常
筋骨格
- 股関節外反による幅広の動揺性歩行、大腿骨頭の無血管性壊死を伴う。
- 末節の骨溶解
- 遠位端の骨溶解を伴う短い鎖骨
- 梨状の胸郭
その他
- 細くて甲高い声
- 低周波伝音性難聴
- 夜間の兎眼(睡眠中に完全に閉眼できない)
臨床診断
5つの主要なカテゴリによって、LMNA関連疾患を定義づけする
カテゴリ1と2はHGPSと定義する。カテゴリ3〜5はHGPSとはしない。
- プロジェリン産生古典的遺伝子型HGPS
- プロジェリン産生非古典的遺伝子型HGPS
- 非プロジェリン産生プロジェリアラミノパチー(鑑別診断を参照)
- プロジェリンを産生しないヘテロ接合性のLMNA 病的ヴァリアント
- 他の遺伝子(例ZMPSTE24)の病的ヴァリアントによる
- 非早老症ラミノパシー(鑑別診断を参照)
- 非ラミノパシー早老症(鑑別診断を参照)
古典的遺伝子型HGPSの診断は発端者の上記の臨床所見と分子遺伝学的検査でヘテロ接合性のLMNA c.1824C>T 病的ヴァリアントが同定されることによる。(参照 表1)
非古典的遺伝子型HGPSの診断は、発端者は、古典的な遺伝子型HGPSに類似した臨床所見を呈し、常染色体優性遺伝で、分子遺伝学的検査でLMNAエクソン11スプライス境界またはイントロン11内のプロジェリンを産生する病的ヴァリアントを有する。(参照表1)

図1 古典的な特徴を呈するHGPSの11歳女児と6歳男児
写真提供:Progeria Research Foundation
分子遺伝子検査のアプローチには、標的遺伝子検査(単一遺伝子検査、複数遺伝子パネル)と包括的なゲノム検査(エクソームシーケンス、ゲノムシーケンス)の組み合わせを含む。
分子遺伝学的検査
単一遺伝子検査
- HGPSの臨床的な所見を有する患者では、最初に、LMNA病的ヴァリアントc.1824C>T(HGPSの90%で同定)の標的解析を実施する。
- 標的解析で病的ヴァリアントが同定されなかった場合、LMNAのシークエンシング解析を実施する。
- 標的解析が未実施な場合、イントロン11を含めたシークエンシグを含めるべきである。
注:LMNAの欠失および/または重複は、HGPSでは報告されていない。
LMNA、ZMPSTE24やその他の関連する遺伝子(参照 鑑別診断)を含む多遺伝子パネルは、症状を説明できない遺伝子の意義不明ヴァリアント(VUS)や病的ヴァリアントの同定において、最も合理的な費用で、疾患の病的ヴァリアントを同定可能である。
注:(1)パネルに含まれる遺伝子と各遺伝子に使用される検査の診断感度は検査室によって異なり、時間の経過とともに変化する可能性がある。(2)一部の複数遺伝子パネルには、このGene Reviewで説明されている疾患に関連しない遺伝子が含まれている場合がある。(3)一部の検査室では、パネルオプションには、個々の検査室設計のパネルおよび/または臨床医によって指定された遺伝子を含む固有の表現型を標的にしたエクソーム解析が含まれる場合がある。(4)パネルで使用される方法には、シークエンシング、欠失/重複分析、および/または他の非シークエンシグが含まれることがある。
包括的なゲノム検査 表現型が早老症の表現型を特徴とする他の多くの遺伝性疾患と鑑別がつかない場合は、包括的なゲノム検査(臨床医が、関与している遺伝子を選定する必要がない)が最良の選択肢となる。エクソームシーケンスが最も一般的に用いられている。ゲノムシーケンシングも可能である。
表1 ハッチンソン-ギルフォードプロジェリア症候群で使用される分子遺伝学的検査
| 遺伝子 | 方法 | 発端者で病的変異が検出される率 |
|---|---|---|
| LMNA | 古典的な遺伝子型c.1824C>Tの標的解析 | 古典的な遺伝子型HGPSの100% |
| すべてのHGPS の~90% | ||
| シーケンス | すべてのHGPSの〜100% | |
| 遺伝子を標的とした欠失/重複解析 | 検出されていない |
- 染色体位置とタンパク質については、遺伝子とデータベースを参照
- この遺伝子で検出された対立遺伝子ヴァリアントついては、分子遺伝学の項を参照
- HGPSの典型的な臨床的特徴を有する患者の約90%は、古典的遺伝子型(ヘテロ接合性 c.1824C>T 病的ヴァリアント)を有する。残りの約10%は、ヘテロ接合性の病的ヴァリアントc.1822G>Aを有する、またはプロジェリン産生するエクソン11またはイントロン11の病的ヴァリアントを有する[ Gordon et al 2018b ]。
- シークエンシングにはイントロン11を含める必要がある。シークエンシング解析は、良性、良性の可能性、意義不明、病的の可能性、または病的のヴァリアントを検出する。病的ヴァリアントには、小さな遺伝子内欠失/挿入およびミスセンス、ナンセンス、およびスプライス部位ヴァリアントを含む。通常、エクソンまたは全遺伝子の欠失/重複は検出されない。
- ディープシーケンスで体細胞モザイクが同定されている。患児は異なる細胞でプロジェリンを産生する2つのヴァリアントを呈した。 [ Bar et al 2017 ]。
- 遺伝子を標的とした欠失/重複解析は、遺伝子内の欠失または重複を検出する。用いられる方法には、定量PCR、長距離PCR、マルチプレックスライゲーション依存プローブ増幅(MLPA)、および単一エクソンの欠失・重複を検出するように設計された遺伝子標的マイクロアレイを含む
臨床的特徴
自然経過
臨床的特徴 古典的および非古典的遺伝子型を有するHGPSは、類似した臨床的特徴と一連の重症度を呈する。
臨床症状
古典的および非古典的遺伝子型ハッチンソン-ギルフォード早老症候群(HGPS)は、小児期に発症し、早老症のいくつかの臨床的症状を特徴とする。早老症の小児は通常、出生時および乳幼児期早期は正常である。
成長障害: 通常、最初の1年間に重度の成長障害が生じる。体重増加不良と皮下脂肪の減少のため、体重は年齢で3パーセンタイル未満になり、体重は身長に比して著明に低下する。身長も年齢の3パーセンタイル未満に低下する。
特徴的な顔貌(図1を参照)には、顔に対して不均衡な大頭、狭い鼻尖と狭い鼻隆起、薄い上下口唇、小さな口、下顎の後退、および小顎が含まれる。弓型(尖塔型)の口蓋円蓋は、罹患した患者の60%〜70%で認める。舌の可動性を制限する短く厚い舌小帯は、罹患した患者の約50%に見られる。狭い気道と硬い喉頭の構造により、甲高い声を生じる。
歯科: 一般的に乳歯の萌出遅延との脱落の遅延を生じる。小さな口、乳歯の脱落遅延、および乳歯の後方からの永久歯の萌出のため、歯叢生が生じる。永久歯の萌出はしばしば部分的である
皮膚: 皮膚所見は出生時には明らかもしれないが、2歳までにすべての患者に認める。硬化性皮膚は、ぴんと張った、肥厚、線維性、硬化、または波状などを呈し、症状や部位はさまざまである。
さらに、下腹部と大腿近位部にくぼみや不規則な小さな盛り上がり(ふくらみ)がみられることがある。また、明・暗斑点、いくつかの丘疹および皮膚の紋様の斑点からなる異常な色素沈着を示す。
毛髪: 部分的な脱毛症は完全な禿頭に進行する。疎なうぶ毛が後頭部に認めることがある。眉毛の喪失は一般的であり、一部にはまつげの喪失を認める。
爪: 指の爪と足指の爪は形成異常がある。
筋骨格: HGPSの患者は、股関節の無血管性壊死(骨壊死)による進行性の股関節外反のために特に股関節脱臼を生じやすい。無血管性壊死はX線で明らかで、股関節痛をおこす。股関節外反は、幅広の動揺性歩行を呈する。それに加え、骨の変化には、末節骨の骨溶解、遠位部の骨吸収による短い鎖骨、洋梨形の胸郭、および年齢に比して低い骨密度などの骨変化を認める。骨折は、HGPSの患者ではあまり一般的に報告されていない。骨格外石灰化は40%の患者に認められるが、臨床的意義は不明である。関節靭帯の張りと骨関節炎による進行性の関節の拘縮の重症度は様々である。
内分泌: 罹患児は性的に成熟をしない。女性は思春期にTanner stage 1(78%)または2(22%)に達し、約60%が初潮を経験する [ Greer et al 2018 ]。妊娠の報告はない。血清レプチン濃度が検出限界以下である。糖尿病が未発症でも、インスリン抵抗性を約50%で認める。
心血管/脳血管: HGPSの患者は通常、脂質代謝に明らかな異常はないが重度のアテローム性動脈硬化症を発症する[ Gordon et al 2005 ]。一般に、血清コレステロール、LDLおよびトリグリセリド濃度は上昇せず、HDL濃度は年齢とともに低下する傾向がある。拡張機能障害は初期の心臓異常であり、通常、組織ドップラー心エコー検査によって5歳すぎに認められる [ Prakash et al 2018]。引き続く心血管機能低下の症状には、心筋の拡張障害とそれに続く心室肥大が含まれる。これは、心臓弁の肥厚や狭窄や不安定な高血圧によって生じる可能性がある。僧帽弁および大動脈弁の異常として石灰化、狭窄、逆流などが、通常、10歳代に発症する。
収縮機能障害は通常、冠動脈不全の有無にかかわらず、疾患の進行によって生じてくる。狭心症、労作時呼吸困難または明白な心不全の臨床症状は、疾患の進行性の経過の後期に認められる。
一過性脳虚血発作、無症候性脳卒中または症候性脳卒中は、早ければ4歳で生じる[ Silvera et al 2013 ]。脳卒中は脳のどの部位でも生じる可能性があるため、さまざまな身体的制限や認知機能の低下につながる可能性がある。部分的および完全な頸動脈閉塞は、プラーク形成から発生する可能性がある。潜在的な血管疾患にもかかわらず、ほとんどの小児は臨床的に脳卒中症状を認めない。
指のレイノー現象は、患者の少数で認める。
死因は、通常、心臓病または脳血管疾患の合併症の結果として生じる。死亡の80%以上は多くは6歳から20歳に心不全および/または心筋梗塞によるものである。平均寿命は約14.5歳[ Gordon et al 2014、Gordon et al 2018a ]。
眼科:夜間の兎眼(睡眠中に完全に閉眼できない)が一般的。その結果、角膜の乾燥や曇りが生じる可能性がある。ドライアイが原因で角膜潰瘍が生じることが少数で認める[ Mantagos et al 2017 ]。
聴覚: 伝音性難聴はすべての年齢層で高頻度に生じ、低周波難聴は高周波よりも多い。 [ Guardiani et al 2011、Gordon et al 2012 ]。
その他:
- 運動と精神の発達は正常。
- 腫瘍の発生率は一般集団よりも増加していない。胸壁の軟骨肉腫で死亡した13歳の患児が1名報告されている[1978 Kingら]。
- 近視または遠視、老人環、老人様の性格の変化、またはアルツハイマー病などの通常の老化に関連する他の変化は報告されていない。
- HGPSの小児の免疫システムは正常である。さまざまな感染症に対して、一般と同様に反応する。創傷治癒は正常。
- 肝臓、腎臓、胃腸、神経および認知機能は正常。
遺伝子型と臨床型の関連
表2 古典的遺伝子型HGPSと非古典的遺伝子型HGPS:原因遺伝子LMNAヴァリアントと臨床表現型との比較
| 遺伝子型 | LMNA病的ヴァリアント (予測されるタンパク変化) |
古典的HGPSと比較した表現型 1 | 患者数 | 文献 |
|---|---|---|---|---|
| 古典型HGPS | c.1824C>T (p.Gly608 =)2 |
脚注3を参照 | 113 | Eriksson et al [2003], De Sandre-Giovannoli et al [2003] |
| 非古典型HGPS | c.1822G>A (p.Gly608Ser) | 中程度 | 5 | Eriksson et al [2003], PRF |
| c.1821G>A (p.Val607 =)2 |
重度、新生児早老症 | 3 | Moulson et al [2007], Reunert et al [2012], PRF |
|
| c.1968G>A (p.Gln656 =)2 |
非常に軽度 | 2 | Hisama et al [2011], Barthélémyetal [2015] |
|
| c.1968 + 1G>C | 重度 | 2 | Iqbal&Iftikhar [2008], PRF |
|
| c.1968 + 1G>A | 重度 | 4 | Moulson et al [2007], Navarro et al [2004], PRF |
|
| c.1968 + 2T>A | 軽度 | 2 | Bar et al [2017], PRF |
|
| c.1968 + 2T>C | 軽度 | 1 | PRF | |
| c.1968 + 5G>A | 非常に軽度 | 2 | Hisama et al [2011], PRF |
|
| c.1968 + 5G>C | 中等度 | 3 | PRF |
HGPS =ハッチンソン-ギルフォード早老症候群; PRF =プロジェリア研究財団の診断テストプログラム
- 古典的遺伝子型HGPSにはさまざまな重症度があり、非古典的遺伝子型HGPSのほとんどの人はその範囲に含まれる。古典的遺伝子型HGPSとの比較は、古典的遺伝子型HGPSの平均的な重症度に基づく。記載されている病的ヴァリアントが、個々でさまざまな重症度の可能性があることに注意する。
- タンパク質レベルの影響が予想されないことを意味する
- LMNA 病的ヴァリアント c.1824C>Tは、表現型が著しく類似している [ Eriksson et al 2003 ]。
浸透率
完全浸透
病名
HGPSは、ハッチンソン-ギルフォード症候群または早老症。
頻度
総人口あたりHGPS児の罹患率は1/2000万[Gordon et al 2014 ]。
HGPSの推定出生率は400万人に1人であり、民族的背景による差はない[ Hennekam 2006 ]。
遺伝学的に関連のある疾患(同一アレル疾患)
LMNAの塩基ヴァリアントを有する約12の明らかに異なる遺伝的状態が同定されている。(OMIM150330)を参照。さらに、LMNAの翻訳後プロセシングに関与する酵素である亜鉛メタロプロテイナーゼをコードするZMPSTE24病的ヴァリアントは、過剰なプレラミンAタンパク質を産生し、関連する表現型を呈する(OMIM 606480)。
非プロゲリン産生早老症ラミノパシーは、古典的および非古典的遺伝子型HGPSと重複する表現型を呈するが、明らかに異なる。Progeria Research Foundation診断テストプログラム、International Progeria Registry、および/またはMedical and Research Database Programを通じて同定されたLMNAのさまざまな病的ヴァリアントは、様々なラミンA異常を産生し、多様な表現型を呈する(鑑別診断を参照)。
非早老症ラミノパシー
異常ラミンAタンパク質を産生する病的ヴァリアントLMNAによって生じる引き起こされる非早老症ラミノパシー:
- 常染色体優性エメリー・ドレフス筋ジストロフィー(AD-EDMD)
- 常染色体劣性エメリー・ドレフス筋ジストロフィー(AR-EDMD)
- 常染色体優性の家族性拡張型心筋症および伝導系の障害(拡張型心筋症を参照)
- 常染色体優性ダニガン型家族性部分型脂肪異栄養症(FPLD)(OMIM 151660)
- 常染色体優性肢帯型筋ジストロフィー1B(LGMD1B)
- 常染色体劣性軸索ニューロパシーシャルコー・マリー・トゥース病2B1(CMT2B1;シャルコー・マリー・トゥース遺伝性ニューロパシーの概要を参照)
- 常染色体劣性下顎骨異形成(MAD)[ Cao&Hegele 2003 ]
- LMNAヴァリアントと特有の臨床表現型を有する症例報告[ Cauxet al 2003、Kirschner et al 2005 ]
鑑別診断
非ラミノパシー早老症 早期老化のいくつかの症状を有する他の症候群:
- 新生児早老症(Wiedemann-Rautenstrauch症候群)(OMIM 264090)
- アクロジェリア(OMIM 201200)
- コケイン症候群
- ハラーマン・ストライフ症候群(OMIM 234100)
- Gerodermiaosteodysplastica(OMIM 231070)
- Berardinelli-Seip先天性脂肪異栄養症(先天性全身性脂肪異栄養症)
- Petty-Laxova-Weidemann早老症(OMIM 612289)
- エーラス・ダンロス症候群、早老症(OMIM 130070)
- ウェルナー症候群
- 下顎骨異形成(遺伝的に関連する障害を参照)(OMIM 248370)
- Nestor-Guillermo症候群(OMIM 614008)
- ペンティネン症候群(OMIM 601812)
- POLR3A関連のWiedemann-Rautenstrauch症候群(POLR3関連白質ジストロフィー、Wambach et al [2018]を参照)
- PYCR1関連のWiedemann-Rautenstrauch様症候群[Lesselet al 2018 ]
臨床的マネジメント
最初の診断時における評価
ハッチンソン-ギルフォード早老症候群(HGPS)と診断された患者の疾患の重症度と必要なことを確定するために、まだ完了していない場合は、以下の評価を推奨する。
- 経過に伴う成長を評価するために、標準の成長曲線に体重と身長をプロットする
- 心電図(ECG)と心エコー
- 心血管系のベースラインの状態を確立するために、頸動脈二重スキャンによる内腔のサイズと内膜の厚さの評価
- 頭頸部のMRI / MRA
- 骨格X線による特徴的な所見の評価:先端骨溶解症、鎖骨吸収、外反股、および骨外の軟部組織の石灰化[ Cleveland et al 2012 ]
- 進行性外反股および/または無血管性壊死の整形外科的評価
- 二重エネルギーX線吸収測定法(DXA)による骨塩密度の評価。注:これは身長年齢に対して正規化する必要あり[ Gordon et al 2011 ]。
- 6分間の歩行テスト、関節の可動域測定、日常生活動作の評価を含む作業療法と理学療法の評価
- 一般的に食事摂取量の減少についての栄養評価
- 聴覚、眼科および歯科の診察
- 臨床遺伝専門医および/または遺伝カウンセラーとの相談
病変に対する治療
完全な臓器別の管理ガイドは、Progeria Research Foundationから入手可能。
標的治療
ロナファルニブはファルネシル化阻害剤である。HGPSの場合、ロナファルニブの標的作用は、HGPSの活性型疾患原因タンパク質であるプロジェリンの翻訳後ファルネシル化を阻害することである。ロナファルニブは経口薬で、初回投与量115mg/m2を1日2回投与し、4ヵ月後には150mg/m2を投与する。毒性(推奨される治療法)は、主に下痢(ロペラミド)、胃腸の不調、吐き気(オンダンセトロン)、食欲不振(シプロヘプタジン)であり、これらは通常、治療開始後数ヵ月で軽減する。ロナファルニブの臨床試験結果から、脈波伝播速度や血管エコー密度から測定される血管拡張能の改善、骨硬直や神経感覚聴力の増加 [Gordon et al 2012]、頭痛の減少 [Ullrich et al 2013]、平均2.5~5年の寿命延長(~20%~30%)[Gordon et al 2014, Gordon et al 2023]が明らかにされている。
支持的治療
成長不全 頻繁な少量の食事は、摂取カロリー量を最大限に高める。
歯科 歯の発達と2重歯列の叢生を避けるために、乳歯の抜歯が必要になることがある。永久歯の萌出遅延とすべての歯が萌出しないかもしれないので、永久歯が完全・ほぼ完全に萌出またはほぼ完全に下降したら、永久歯萌出のためのスペース確保のため、乳歯を抜歯しなければならない。乳歯が抜歯されると、永久歯は経過とともに正規の位置に移動することがよくある。
皮膚 屋外での活動には、頭部含むすべての皮膚の露出部に日焼け止めを使用することを推奨する。
整形外科 可能な場合は理学療法とボディブレースによる股関節脱臼の保守的な管理を行う。外科的矯正が必要な場合、挿管と麻酔のガイドラインは特別な注意を要する。外科的手術は、脳卒中および/または心臓イベントの実質的なリスクが高くなると考える。体脂肪の不足から足の疼痛が生じるので、足を評価し、靴の中敷きが必要かどうかを判断する。骨折率は一般的な小児人口と同等である。骨折時の治療と治癒は通常通りである。
大小の関節の可動域を維持するために、定期的な理学療法と作業療法が推奨される。
積極的なストレッチと水中運動による強化が推奨される。
機能障害の膣から月経や小出血がある場合、子宮内膜を安定させるために、短期(3〜6か月)の低用量の経口避妊薬が有用である。
心血管 脂質プロファイルが異常にならない限り、通常の健康的な食事が必要である。異常になった時点で、運動、食事の調整および必要に応じた投薬を含む治療を行う。特により進行した心血管疾患のある患者にとって、貧血、脱水症、高熱を避けることは重要である。適切な経口での水分補給を維持することを勧める。
心血管または神経学的状態(脳卒中、狭心症または心臓発作に起因する)が低下する前に、関節可動域の制限および変形性関節症や股関節脱臼による限界を考慮して、小児は許容できる範囲で身体的な活動するように奨励する。
血管閉塞、一過性脳虚血発作、脳卒中、狭心症または心筋梗塞が生じた際は、通常、推奨されるアスピリン以外の抗凝固薬(二次合併症の予防を参照)も必要に応じる。
脂質代謝は通常正常だが、異常を呈した場合は食事療法±スタチン療法を実施する。
薬 投与量は、年齢ではなく、体重または体表面積に基づく必要がある。麻酔薬は特に注意して使用する。
- 狭心症が発症した場合、ニトログリセリンはしばしば有効である。
- うっ血性心不全がある場合は、定期的な抗うっ血療法を実施する。
全身麻酔と挿管は、理想的には気管支ファイバー挿管で、細心の注意で実施する必要がある。HGPSの患者は、下顎後退、硬化した喉頭構造、および狭く形態異常のある気道を有する。さらに、血管硬化による血圧の変化に対して極めて高い感受性を示す場合がある。
眼科 眼科医によって角膜の乾燥、曇り、または潰瘍形成は十分評価する。ドライアイについては、日中は眼の潤滑剤、睡眠中は保湿軟膏またはスキンテープで眼瞼を閉じることにより治療する。
難聴 低周波の伝音性難聴は、日常生活の活動中の動作をほとんど妨げない。教室の前方の座席にすることは有用である。臨床的に必要な場合は、補聴器を装用する。
教育 知性と成熟度は正常であり、通常、年齢に応じた学校教育が必要である。
感染症は通常、一般の小児と同様に治療する。
二次病変の予防
アスピリン 成人の研究において、低用量のアスピリンが心臓発作や脳卒中の予防に有用であることから、HGPSの小児に、低用量アスピリン療法 1日あたり2〜3 mg / kgの用量で投与することが適切である。注:地域で水痘またはインフルエンザが流行している場合、ライ症候群のリスクが高まるため、その期間中はアスピリンを中止することをすすめる。
適切な水分補給 血管系は一般的に柔軟性が低くなり、血管の代償性の低下により脳卒中や心臓合併症のリスクが経年的に高くなるので、適切な経口補水を勧める。これは、暑い時期や飛行機での旅行中に特に重要である。
ビタミン補給 市販の日用マルチビタミン錠剤の標準量が適切である。骨外石灰化沈着の悪化、血管内プラーク沈着を進行させる可能性があるため、カルシウムの補給は推奨されない。食事を通して、正常なカルシウムレベルの維持が奨励される。
フッ化物サプリメントは、適宜、推奨される。
予防接種 すべての予防接種は、通常の投与量と投与スケジュールが推奨される。予防接種は通常、一般と同様である。
経過観察
完全な臓器別管理ガイドは、Progeria Research Foundationから入手可能。
毎年または半年ごと 心血管疾患の管理のため、適切なサイズのカフによる血圧測定、ECG、心エコー図、および頸部二重スキャン。注:小児は、重大なECGの変化が出現する前に、重度の頸動脈アテローム性動脈閉塞を生じる可能性がある。
毎年
- 頭痛と脳卒中の兆候や症状の神経学的評価
- 頭頸部のMRI / MRA 経年的にリスクが高まる血管の変化と潜在的梗塞を評価
- 脂質代謝
- 歯科検診、レントゲン、清掃
- 整形外科的評価 股関節の無血管性壊死(骨壊死)および進行性の股関節外反による乗馬の姿勢および潜在的な股関節脱臼の可能性
- 6分間の歩行テスト、関節の可動域測定、日常生活動作の評価を含む作業療法および理学療法の評価
- ドライアイに特別な注意を要する眼科検査
- 低周波の伝音性難聴に特別な注意を要する聴覚の評価
回避すべき薬物や環境
怪我のリスクが高まるため、小児患者は、背がとても高く、体格が大きな人達らの大勢の真中にいることを避ける。身体活動は自己で制限をする。トランポリンやバウンスハウス(エアー遊具)など表面が一様でないものは、股関節形成不全を悪化させるので避ける。年下の子供に担ぎあげられることは避ける。脳卒中のリスクが高まるため、脱水症状を避ける。
リスクのある親族の検査
リスクのある親族の検査に関連する遺伝カウンセリングは、遺伝カウンセリングの項を参照。
研究中の治療法
HGPSまたは早老症の治療研究は、米国ではClinicalTrials.gov、ヨーロッパではHGPSの臨床試験に関する情報については、EU Clinical Trial Registerを検索する。
現在HGPSのヒト臨床試験調査中の2つの治療法は、ロナファルニブとエベロリムスである(図2を参照)。現在、2つの臨床試験があり、1つはロナファルニブ単剤療法を投与、もう1つはロナファルニブとエベロリムスの併用療法である。
図2. 現在調査中のHGPSの臨床治療試験での翻訳後修飾と薬剤。
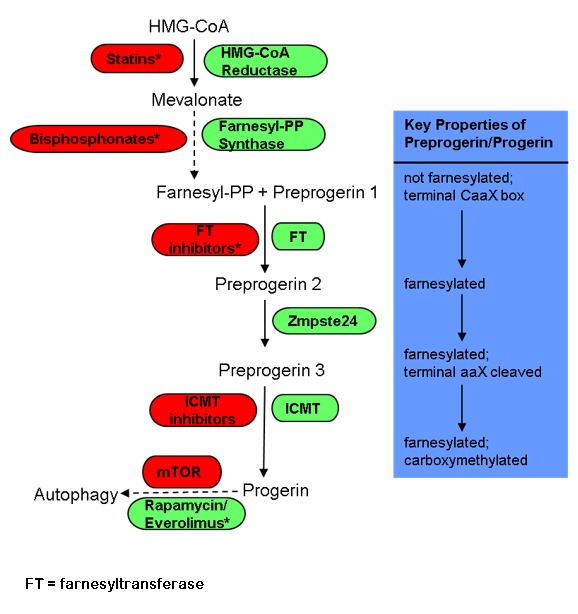
緑の項目はパスウエイを促進、赤の項目は経路を阻害する。*は臨床試験でテストされている薬剤。エベロリムスはmTORを阻害するラパマイシンアナログで細胞のオートファジーを促進する。
- ロナファルニブは、治験中のファルネシルトランスフェラーゼ阻害剤である。HGPSにおいて、その標的作用は、HGPSの活性化している疾患原因タンパク質であるプロジェリンの翻訳後ファルネシル化を阻害する。ロナファルニブは、1日2回経口投与される薬。ロナファルニブの臨床試験結果は、体重増加率、脈波伝播速度と血管エコー密度で測定された血管伸展性、骨の硬さ、感覚神経聴力[ Gordon et al 2012 ]、頭痛[ Ullrich et al 2013 ]、および寿命を改善した[ Gordon et al 2014、Gordon et al 2018b ]。
- エベロリムスは、細胞のオートファジーをさらに活性化するラパログ(ラパマイシン様薬物)mTOR阻害剤である。1日1回経口投与薬。エベロリムスは、非HGPSの治療薬として承認されている。ラパマイシンは、オートファジーの活性化を介してHGPS線維芽細胞の細胞表現型を改善し[ Cao et al 2011、Cenni et al 2011 ]、ラミンA欠損マウスモデルの寿命を延長した。臨床試験の結果は未公表である。
その他
- プラバスタチンおよびゾレドロネートと組み合わせてロナファルニブを投与する臨床治療試験は、骨塩密度の増加を認めたが、ロナファルニブ単剤療法よりも他の改善はなかった[ Gordon et al 2016 ]。
- プラバスタチンとゾレドロネートの併用療法の臨床治療試験は実施中である。
以下の申請された治療法は、ヒトでは実施されていない(表3を参照)。これら治療法の可能性の根拠についての包括的なレビューについては、Strandgren et al [2017]を参照。
表 3 In vitroおよび/またはマウス研究でのみ試験された治療
| 治療薬 | Pathway | Target |
|---|---|---|
| All-trans retinoic acid | オートファジー | Progerin 代謝 |
| アンチセンスオリゴヌクレオチド | スプライシング機構制御 | Lamin C / prelamin A スプライシング &/or異常 LMNA スプライシング |
| DOT1L 阻害剤 | 細胞初期化 | DOT1L |
| Isoprenylcysteine carboxyl methyltransferase (ICMT) knock-down = shICMT | Prelamin A 翻訳後修飾 | ICMT |
| 遺伝子編集 | CRISPR/Cas9 | LMNA 塩基配列 |
| JH4 | Progerin-lamin A/C 結合 | Progerin-lamin A/C 結合 |
| メトホルミン | AMPK活性化 | 肝臓糖代謝調節機構 |
| メチレンブルー | ミトコンドリア生合成 | ミトコンドリア機能 |
| MG132 (プロテアソーム阻害剤 ) | オートファジー | Progerin 代謝 |
| Mono-aminopyrimidines | Prelamin A 翻訳後修飾 | Prelamin A ファルネシル化 |
| N-アセチルシスチン | 酸化ストレス | 活性酸素 |
| caNRF2 | NRF2 再活性 | NRF2 |
| OSKM 誘導 | エピジェネティック リモデリング | 一部細胞の初期化 |
| ピロリン酸 | 細胞外石灰化代謝 | リン酸カルシウム沈着 |
| ラパマイシンおよび類似体 | オートファジー | Progerin 代謝 |
| Remodelin | 微小管 | NAT10 |
| Resveratrol | SIRT1 活性 | SIRT1 |
| サリチル酸ナトリウム | NF-κB シグナリング | NF-κB 阻害 |
| 幹細胞移植 | 幹細胞機能 | 組織再生 |
| Sulforaphane | オートファジー | Progerin 代謝 |
| テロメラーゼ | テロメア長 | テロメア 1 |
| Vitamin D | Vitamin D レセプター シグナリング | Vitamin D 受容体 |
- Li et al [2017]、Strandgren et al [2017]には含まれていない
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
ハッチンソン-ギルフォード早老症候群(HGPS)は通常、常染色体優性遺伝の新生突然病的ヴァリアントによって生じる。
家族構成員のリスク
発端者の両親
- ほとんどすべてのHGPSは、新規の病的ヴァリアントの結果で生じた疾患である。
- 現在、生存している古典的HGPS遺伝子型を有する患者のうち、Progeria Research Foundation Diagnostics Programによって、親の明らかな生殖細胞系列(または体細胞および生殖細胞系列)モザイク(約3%)を同定した [ Wuyts et al 2005 ]。
- 発端者の親は非罹患。
発端者の同胞
- HGPSは通常、新規の病的ヴァリアントによって生じるため、発端者同胞の再発リスクは低い。
- 現在、古典的HGPS症例と診断された113名中のうち双胎ではない同胞症例が3名(2%)同定されている。113名中のうち同胞の非罹患数は不明。したがって、遺伝的に診断されたHGPSでは、その後の妊娠の再発リスクは、一般集団の発生率400万人に1人よりも有意に高いが、それでも再発率は低い。
発端者の子
古典的および非古典的なHGPSの生殖についての報告はない。
関連する遺伝カウンセリング上の諸事項
明らかに新生突然ヴァリアントによる家族
新生病的ヴァリアントの由来 評価された家族数は少ないが、ある研究では4/4例が父親由来であった[ Eriksson et al 2003 ]。Wuyts et al [2005]によって報告されたモザイク症例は、病的ヴァリアントは母親由来であった。父親の年齢効果に関する研究では、父親の年齢が平均して約5才、有意に加齢していた [ Brown et al 1985 ]。近親婚の増加はない。
リスクのある無症状の家族に対する検査
家族計画
DNAバンキング
DNAバンキングは、将来の使用に備えてDNA(通常は白血球から抽出)を保管することである。検査方法と遺伝子、対立遺伝子のヴァリアントおよび疾患の理解は,将来進展する可能性があり、罹患患者のDNAの保管について考慮する必要がある。
出生前診断
出生前検査と着床前遺伝子検査
家族内にLMNA 病的ヴァリアントが同定された患者がいる場合、再発リスクの増加(親の生殖細胞系列モザイクの稀な可能性)のため、出生前検査および着床前遺伝子検査は可能。
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A ハッチンソン-ギルフォードプロジェリア症候群:遺伝子とデータベース
データは、次の標準参照から編集。
遺伝子HGNC;染色体遺伝子座OMIM;タンパク質 UniProt。リンクが提供されているデータベース(Locus Specific、HGMD、ClinVar
表B ハッチンソン-ギルフォード プロジェリア症候群のOMIMエントリー(OMIMですべて表示)
| 150330 | LAMIN A/C; LMNA |
| 176670 | HUTCHINSON-GILFORD PROGERIA SYNDROME; HGPS |
疾患の分子基盤
MNA 病的ヴァリアンアトc.1824C>T、CからTへのヴァリアントは、アミノ酸翻訳グリシンの変更はないが、潜在的スプライス部位を活性化し、エクソン11の3'部分に150塩基対の欠失を有する転写産物をもたらす。この選択されたmRNAの翻訳後修飾ではプロジェリンとC末端近くの50アミノ酸欠失がある異常なプレラミンAタンパク質を生成する。50アミノ酸の欠失は、各細胞分裂での核膜の解離と再融合に関与するリン酸化部位とともに、プレラミンAの末端18アミノ酸のタンパク質分解的切断をもたらす認識部位も失う。
HGPSの疾患の重要な要素は、おそらく持続的なプロジェリンのファルネシル化であり、これにより、プロジェリンは核内膜に恒久的に挿入され、細胞の老化とともに蓄積し、細胞に徐々に大きな損傷を与える可能性がある。ファルネシル基の除去の失敗が、HGPSでみられる表現型の少なくとも一部の原因となりうることは、非ファルネシル化プロジェリンを合成するように設計された細胞、またはファルネシル化阻害剤によって非ファルネシル化プロジェリンを産生するマウスモデルの研究によって強く示唆されている。
プロジェリンタンパク質の産生をもたらさない他のLMNAヴァリアントは、さまざまな構造と機能(核膜ラミン関連タンパク質との相互作用を含む)異常を有するラミンAタンパク質を産生し、これらはすべて、いくつかの観点ではHGPSと重複するさまざまな表現型を呈する細胞および臓器の病態を生じさせる。
遺伝子構造 LMNAの翻訳領域は約24kb、12個のエクソンを含む。遺伝子およびタンパク質情報の詳細な要約については、Table A、遺伝子を参照。
病的ヴァリアント 表 4を参照。
表4このGeneReviewで議論されたLMNA遺伝子の病的ヴァリアント
| DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | 参照配列 |
|---|---|---|
| c.1821G>A 1 | p.Val607 = 2 | NM_170707 .2 NP_733821 .1 |
| c.1822G>A | p.Gly608Ser | |
| c.1824C>T 1 | p.Gly608 = 2 | |
| c.1968G>A | p.Gln656 = 2 | |
| c.1968 + 1G>A | (スプライスドナーサイトヴァリアント) | |
| c.1968 + 1G>C | (スプライスドナーサイトヴァリアント) | |
| c.1968 + 2T>A | (スプライスドナーサイトヴァリアント) | |
| c.1968 + 2T>C | (スプライスドナーサイトヴァリアント) | |
| c.1968 + 5G>C | (スプライスドナーサイトヴァリアント) | |
| c.1968 + 5G>A | (スプライスドナーサイトヴァリアント) |
表にリストされているヴァリアントは、作成者によって提供されており、Gene Reviewsのスタッフは、ヴァアントの分類を独自に検証していない。
Gene Reviewsは、ヒトゲノムヴァリアント学会(varnomen .hgvs.org)の標準的な命名規則に従っている。命名法の説明については、クイックリファレンスを参照。
1.インフレームエクソン11潜在的スプライス部位活性化ヴァリアント体
2.=タンパク質レベルへの影響が予想されないことを示す。
正常な遺伝子産物 核ラミンは、核膜の内膜を裏打ちするタンパク質含有層である。哺乳類では、それはポリペプチドのファミリーで構成されており、主成分はラミンA、B1、B2、およびCであり、分子量は60,000〜78,000。ラミンAおよびCは、LMNA / C遺伝子転写産物の選択的スプライシングによって生成される。エキソン10内でのスプライシングはラミンCを生じ、12個すべてのエキソンの転写はラミンAを産生する。ラミンB1とB2は別々の遺伝子によってコードされ、ラミンB1とB2で、既知の早老症の病的ヴァリアントは報告されていない。
ラミンAは通常、前駆体分子(プレラミンA)として生成され、4つの主要な翻訳後修飾ステップを経る。まず、プレラミンAのカルボキシル末端にCAAX(システイン/脂肪族アミノ酸/脂肪族アミノ酸/任意のアミノ酸)ボックスが含まれ、ファルネシル化によって修飾される。ファルネシル化に続いて、最後の3つのアミノ酸の切断、C末端のメチル化、および内部タンパク質分解切断が生じる。CAAXボックスおよびファルネシル基とともに最後の15個の翻訳されたアミノ酸を除去すると、646個のアミノ酸を持つ成熟ラミンAが生成される。
異常な遺伝子産物 LMNAのコドン608のHGPSを引き起こすヴァリアントは、エクソン11内の潜在的なスプライス部位の活性化を引き起こし、C末端近くに50アミノ酸を欠くプレラミンAの生成をもたらす[ Eriksson et al 2003 ]。c.1824C>T病的ヴァリアントによりスプライシング異常が生じ、ファルネシル化されたままCAAXボックス有するプレラミンAが生成され、翻訳後修飾の最終ステップで通常生じる、ファルネシル基残基とそれにつながる最後の16アミノ酸からなるエンドプロテプロテアーゼ分解切断部位が欠失している。その結果生じた、ファルネシル化された短縮タンパク質は、プロジェリンと呼ばれる。脂溶性ファルネシル基は、プレラミン(プロジェリン)を核内膜に結合し、ファルネシル化タンパクは切断されずに、核内膜との長期的なプロジェリンの接着をもたらすことになる。
更新履歴:
-
GeneReviews著者: Leslie B Gordon, MD, PhD, W Ted Brown, MD, PhD, and Francis S Collins, MD, PhD
日本語訳者:小崎 里華(国立成育医療研究センター 遺伝診療科)
GeneReviews最終更新日: 2019.1.17 日本語訳最終更新日: 2021.1.13. - Gene Reviews著者: Leslie B Gordon, MD, PhD, W Ted Brown, MD, PhD, and Francis S Collins, MD, PhD.
日本語訳者:小崎 里華(国立成育医療研究センター 遺伝診療科) GeneReviews最終更新日: 2023.10.19. 日本語訳最終更新日:2024.10.3. [in present]
原文: Hutchinson―Gilford Progeria syndrome
| X POST |
![]()