筋強直性ジストロフィー2型
(Myotonic Dystrophy Type 2 (DM2))
Gene Reviews著者: Joline C Dalton, MS; Laura PW Ranum, PhD; John W Day, MD, PhD
日本語訳者: 堀江 里歩、(監訳) 高橋 正紀(大阪大学大学院医学系研究科保健学専攻 機能診断科学講座)
Gene Reviews 最終更新日:2013.7. 3. 日本語訳最終更新日: 2020.01.05.
要約
疾患の特徴
筋強直性ジストロフィー2型(DM2)は筋強直現象(90%の患者でみられる)、骨格筋機能低下(筋力低下、筋痛、こわばり:82%の患者でみられる)、より低頻度ではあるが心伝導異常、虹色の後嚢下白内障、インスリン抵抗性2型糖尿病、精巣不全を特徴とする。筋強直現象(弛緩遅延をともなう不随意の筋収縮)は10歳以前にみられると報告されているが、通常発症は20歳代で変動するあるいは一時的な筋痛、頸部や手指の屈筋の筋力低下で発症する。それに引き続き、肘関節の伸筋、股関節の屈筋、伸筋の筋力が低下する。顔面筋や足首の背屈筋の筋力低下はより少ない。筋強直現象が重い症状となることは稀である。
診断・検査
CNBP(ZNF9)遺伝子は筋強直性ジストロフィー2型と関連する唯一の遺伝子である.CNBP遺伝子のイントロン1は複雑なリピートモチーフ(TG)n(TCTG)n(CCTG)nを持つ。CCTGリピートの伸長がDM2を引き起こす。伸長したアレルではCCTGリピート数は約75から11000以上であり、平均すると約5000である。CNBP遺伝子のCCTGリピートの検出率は、通常のPCR法、サザンブロット解析、PCRリピートプライムドアッセイを組み合わせると、99%以上となる。
臨床的マネジメント
症状の治療:
筋力低下に対する必要性に合わせて短下肢装具・車いすその他の装具、不整脈のある患者では除細動器の埋め込み、視力に影響を及ぼすレベルの白内障の切除、男性の性腺機能低下症に対するテストステロン補充。筋強直現象が治療を要することは少ない。通常の身体活動は筋力の維持や筋痛のコントロールに役立つと考えられる。筋痛に対して一定の効果が報告されている薬剤にはメキシレチン、ガバペンチン、非ステロイド系消炎鎮痛薬、低用量甲状腺ホルモン補充、低用量ステロイド(5mgプレドニゾロン隔日など)、三環系抗うつ薬がある。
二次的合併症の予防:
麻酔によるリスクが高まる可能性があるため、手術前後の心機能及び呼吸機能の評価、また、二次的な筋力低下を軽減するための甲状腺機能低下症に対する迅速な治療が推奨される。
経過観察:
心伝導障害および心筋症の検索・経過観察のため年一回の心電図、心エコー検査あるいできれば心臓MRI、年一回の空腹時血糖、HbA1c測定、男性患者に対する二、三年に一度の性腺機能低下症に対する検査である。
避けるべき薬剤・状況:
筋力低下の増強と関連付けられた場合のコレステロール低下薬。
遺伝カウンセリング
筋強直性ジストロフィー2型は常染色体優性遺伝である.今のところ、生物学的両親の分子遺伝学的検査を行ったすべての患者で一方の親がCNBP遺伝子のリピート伸長をもっており、新規突然変異は報告されていない。CNBP遺伝子のリピート伸長をもつ個人(患者)の子は、それぞれ50%の確率でリピート伸長を受け継ぐ。異常アレルの大きさや検出可能な異なるリピート伸長の合計から病気の程度や発症年齢、臨床症状を予測することはできない。CNBP遺伝子のリピート伸長の存在がどちらかの親で特定されていれば,リスクのある妊娠に対する出生前診断は可能である.
診断
臨床診断
次の臨床所見があるとき、筋強直性ジストロフィー2型(DM2)を疑う。
- 筋力低下:病初期には徒手筋力テストにて認められる頸部屈筋、手指屈筋の筋力低下。進行期には筋力低下はしばしば殿筋、腰帯筋におよび、階段の上りや椅子からの立ち上がりが困難となる。
- 筋強直現象(持続的筋収縮):10歳以前に把握ミオトニー(強く握ったこぶしを素早く開けない現象)、叩打ミオトニー(ハンマーで筋を叩打した後に現れる筋の持続的収縮)、あるいはミオトニー放電(筋電図でみられる反復性自発性放電)として観察される。しかしDM2患者におけるミオトニーは筋電図で常に検出できるとは限らない。
- 後嚢下白内障:直接検眼鏡検査において非特異的な小胞や混濁として、細隙灯顕微鏡検査において赤色あるいは緑色の特徴的な後嚢下虹色混濁として認められる。
- 心伝導障害、進行性心筋症:心伝導障害は通常の心電図検査で房室ブロックや様々な心室内伝導障害として、進行性心筋症は心エコー検査で拡張型心筋症として認められる。
- 低γグロブリン血症:血清蛋白電気泳動におけるγグロブリン分画の減少や免疫電気泳動におけるIgG, IgMの低下として認められる。これらは筋強直性ジストロフィー症1型、2型患者の75%で認められるが、臨床的異常には関連しない。
- インスリン抵抗性:血清中のインスリン濃度は正常あるいは上昇しているのにもかかわらず、ブドウ糖負荷試験において血糖値の回復が障害される。これにより高血糖、糖尿病がおこりやすくなる。
- 男性の原発性性腺機能不全:血清のテストステロン値の低下、FSH値の上昇、精子減少症、および不妊がみられる。
検査
筋生検 筋線維の萎縮、濃縮した核(pyknotic nucleus)をもった高度に萎縮した筋線維の散在、中心核を持つ筋線維の著明な増加などがみられる。これらの所見は筋強直性ジストロフィー1型(DM1)と共通しており、これらの所見から病理学的に両者を鑑別することはできない。
- タイプ1線維の萎縮は先天性のDM1でよくみられ、DM2と区別される。
- タイプ2線維優位の筋萎縮はDM2でよくみられる。
分子遺伝学的検査
遺伝子 DM2に関連しているとされているのはcellular nucleic acid-binding protein (zinc finger protein 9)蛋白をコードするCNBP(ZNF9)遺伝子のみである。CNBP遺伝子のintron 1は複雑なリピートモチーフ(TG)n(TCTG)n(CCTG)nを持つ。CCTGリピートの伸長がDM2を引き起こす。
アレルのサイズ
正常アレル リピートを構成する3つの要素(TG, TCTG, CCTG)のくみあわせは、正常アレルにおいても、異常アレルにおいても存在する。また正常アレルのCCTGリピート配列では、1個以上のCCTGと異なる4塩基(TCTGあるいはGCTG)による中断がみられる。正常アレルの全体のリピート部分の長さは104-176塩基対である。TGとTCTGリピート数は非常にばらつきが大きいので、アレルの大きさは、塩基配列決定による正確なCCTGリピート数ではなく、通常の分子遺伝学的検査により決定されるこの部位全体の塩基数で報告される。
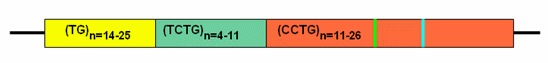
図1:DM2における複雑リピート構造。DM2のリピート構造はTG, TCTGおよびCCTGをこの順序で含む複合体である。各リピートの長さはさまざまである。
正常のアレルではCCTG部分は一般に11~26の4塩基リピートを持ち、典型的には0から2個の4塩基による中断(図では||の縦線で示されている)をCCTGリピート内あるいはモチーフ内に持つが、多くはCCTGリピート部分にGCTCやTCTGによる中断が別々に存在する。
病原性アレルではCCTGリピート部分のみが中断なく伸長し、全体のリピート長が75~11000である純粋なCCTGリピートとなる。
24の正常アレルの塩基配列を解析した研究からは以下のようなことが分かっている。
- 中断されないCCTGリピート数は最高9である。(ただし変異がおきやすい正常アレルという例外(後述)もある)
- 正常におけるGCTGやTCTGの中断を含んだCCTGリピートの長さは11~26リピートである。
- 正常人の85%では2つのアレルで複合したリピート部分の長さが明らかに異なり、PCR解析でその違いが検出される。
変異がおきやすい正常アレル(中間あるいは前変異アレルともいう) 177~372塩基対(27~74CCTGリピートに相当する)の大きさのCNBP遺伝子アレルは報告されていない。さらに、正常アレルのうちで、ある大きさの範囲にあることや特別な塩基配列をもつことが、病的な大きさへの伸長と関連があるのかは、不明である。正常アレルのCCTG配列の中に通常見出される塩基配列の中断はCNBPアレルの病的なCCTG伸長の中に見つけることができない。正常アレルにあるこうした中断がないことが、次の世代において不安定性を増し、リピート伸長しやすい傾向があるのかもしれない。こうした不安定性は他の疾患で確認されているが、DM2では今のところみられていない。しかしながらこの仮説に一致して、すべての患者で認められるものと同じハプロタイプ上に中断のない20CCTGリピートを認める非発症者が報告された。この人のリピートは子供の世代への継代過程では伸長しなかったが、将来の世代では中断のないことが伸長につながる可能性もある。正常の中でもより長いCNBPリピートが病的な範囲に伸長することは確認されていないので、177-372塩基対は、正確には前変異というより境界域とされる。
完全浸透(異常)アレル 塩基配列を調べられたより短い病的アレルの中で、複合リピートのCCTG部分だけが伸長することが報告されている。より大きな伸長では、正確に塩基配列を調べることができないが、372塩基対(75CCTGリピートに相当する)以上のアレルは完全に浸透し、DM2の症状を引き起こす。病的なアレルの大きさは372から44000以上の塩基対(75-11000CTGリピートに相当する)におよび平均は約20000塩基対(約5000リピート)である。
CCTGリピートには以下のような性質がある。
- 体細胞不安定性 CCTGリピートは年齢によって増大する。患者の25%以上は末梢血中に検出される二つ以上の長さのCCTG伸長を持つ。この体細胞におけるCCTGリピートの不均一さのため病的な閾値の確立は困難である。例えば一つのアレルで検出される最も小さいCCTGリピート伸長(約75CCTGリピートあるいは約300塩基対)を持つ患者が、11000CCTGリピートあるいは44000塩基対以上の非常に長いCCTGリピート伸長を含んだアレルを持つこともある。この伸長したアレルの片方あるいは両方が病気をおこしえる。
- 世代間の不安定性 次の世代への遺伝において、CNBP遺伝子リピート長は急激に減少することがある。父親、母親どちらから遺伝するかによって、この減少の程度に明らかな差異は認められない。しかしながら著明な体細胞不安定性とリピート長の年齢による増大があるため、この現象の解釈は複雑である。
臨床的検査
病原性変異体のターゲット解析
CCTG伸長のサイズが大きいこと、体細胞不安定性(CCTG伸長の体細胞不安定性によるアレル長の変化)が存在することから異常なCNBPアレルの検出は困難である。CNBPのCCTGリピート検出率は、通常のPCR法、サザンブロット解析、PCRリピートプライムドアッセイの組み合わせにより99%以上となる。2012年にDM1とDM2の分子診断に関する診療ガイドラインと推奨が発表された。
表1.筋強直性ジストロフィー2型の診断のための分子遺伝学的検査法
| 遺伝子1 | 検査方法 | 変異の検出>2 | 検査法による 変異検出頻度3 |
|---|---|---|---|
| CNBP | 病原性変異体のターゲット解析4 | イントロン1でのCCTG4塩基リピート伸長 | 99%5 |
| 塩基配列解析6 | 塩基配列変異 | 報告症例なし7 |
- 表Aを参照。染色体の遺伝子座とタンパク質の遺伝子とデータベース。
- アレル変異に関しては分子遺伝学を参照。
- 標的となる遺伝子に存在する変異を検出するために用いる検査方法の能力
- 以下の項目が関連する可能性がある
- 通常のPCR分析ではリピートの伸長を増幅することができないため、正常サイズのアレルは検出できるが、異常サイズのアレルは検出できない。
- サザンブロット解析はリピート伸長の約80%を検出できる。
- PCRリピートアッセイはCCTGリピート増幅の検出を容易にするために開発された。このアッセイでは、伸長したCCTGリピート近接部位およびリピート内にプライマーを作ることによって、伸長したアレルはさまざまなリピートサイズを持つスミアとして、正常アレルは個別のバンドとして検出することができる。PCRリピートアッセイ産物は特異性を確保するため、内部プローブで検出する必要がある。
- 検出頻度は検査法により異なる。通常のPCR解析、サザンブロット解析、PCRリピートアッセイを組み合わせて行った場合、変異検出率は99%以上となる。
- 塩基配列解析は、良性、おそらく良性、重要性不確実、おそらく病原性、病原性のいずれかであるかを検出する。病原性変異体には、小さな遺伝子内の欠失・挿入、ミスセンス、ナンセンス、およびスプライス部位の変異体が含まれる場合がある。通常、エクソンまたは全遺伝子の欠失・重複は検出されない。
- 近接するTG,TCTGリピートの大きさの多様性、CCTGリピート中にある中断数の多様性のため、CCTGリピート数は塩基配列解析により決めなくてはならない。患者におけるリピート複合の中のCCTG部分のみが伸長していることを示すためには、いくつかの伸長したアレルの塩基配列を決めることが必要であったが、多くの伸長部分は塩基配列を決めるための反応を行うには長すぎるため、塩基配列解析による正確なCCTGリピート長決定は、診断目的としては有用ではない。
検査結果の解釈
二つの正常サイズアレルが明確に分離できる場合は、PCR解析だけでDM2の診断を除外することができる。
ゲノムDNAのサザンブロット解析を行うことにより、80%以上の患者でアレルの伸長を検出できるが、CCTG伸長の体細胞不安定性および近接する病的ではないリピートの大きさの多様性のため、伸長の大きさは必然的に推測となる。高度の体細胞モザイク現象のため、様々な長さのアレルは電気泳動中に共移動せず、検出が困難になり、サザンブロット解析では伸長の約20%を検出することができない。。
PCRリピートプライムドアッセイは病的範囲に延長したアレルの存在を確認できるが、全体の伸長の長さを決定することはできない。
検査手順
発端者の診断の確認/確立 通常のPCR解析で一つのバンドしか見られなかった場合、一つの正常サイズアレルの存在(正常者の15%およびすべてのDM2患者)を意味し、被検者は同じ大きさの正常サイズアレルをもつのか、伸長したアレルを持ちその長さのためにPCRによって増幅できないのかを区別するために、サザンブロット解析とPCRリピートプライムドアッセイの両者を行う必要がある。
予測検
リスクのある無症候性の成人家族に対して検査を行う場合には、家族の病原性変異を事前に特定しておく必要がある。
出生前診断および着床前遺伝子診断(PGD)
アットリスクの妊娠に対する検査を行う場合には、家族の病原性変異を事前に特定しておく必要がある。
臨床所見
臨床症状
筋強直性ジストロフィー2型(DM2)は筋強直現象(90%)、骨格筋機能不全(筋力低下、筋痛、及び筋のこわばり)(82%)、および以下に示すような一見無関係な臨床的特徴の一貫した組み合わせによって特徴づけられる:心伝導異常(19%)、虹色の後嚢下白内障(36%-78%、年齢とともに増加)、およびインスリン抵抗性(25%-75%、年齢とともに増加)、精巣機能不全(29-65%)を含む一連の特異な内分泌異常。
DM2患者は20歳台で、最も一般的な症状である筋力低下や筋痛で発症することが多い。しかしながら10歳以前に筋強直現象が見られることも報告されている。筋強直性ジストロフィー1型では成人後に変性疾患として発症したり、乳児期あるいは小児期に様々な重症先天型として発症することがあるのに対し、DM2は発育異常とは無関係であり、小児期には重い症状は示さない。DM2家系のどの患者も発育障害を示さないということは、信頼性があり、臨床的に重要な、DMの二つの病型の違いである。
骨格筋機能低下
DM2患者は筋力低下、筋痛、筋強直現象のためしばしば医療対象となる。病初期には頸部屈筋と手指屈筋が障害される。続いて筋力低下は肘関節伸筋、股関節屈筋、伸筋においてもみられるようになる。30%の患者では股関節筋の筋力低下は50歳以降で出現する。
顔面筋と足関節の背屈筋の筋力低下がみられることもあるが、頻度はより少ない。
筋強直現象、(すなわち筋肉の易興奮性による不随意の筋収縮および弛緩の遅延)はほとんどすべてのDM2患者で認められるが、ごく一部の患者でしか重度の症状を呈さない。
変動するあるいは一過性の筋痛は大多数の患者で報告されており、それにより患者が消耗する場合がある。
多臓器疾患としての特徴
虹色の後嚢下白内障は細隙灯検査で早ければ10歳代に認められる。報告されている白内障摘出年齢は28から74歳である。
DM2患者での心症状はDM1より軽症のようであるが、DM2患者では房室や心室内の伝導ブロック、不整脈、心筋症、突然死と関連する可能性がある。ある先行研究では、DM2ではもともと考えられた以上に左心室の機能障害と関連していることが示された。
麻酔の合併症はDM2患者では報告されておらず、おそらくDM1より合併症の頻度は低い。DM1では手術中および手術後の不整脈、呼吸抑制および不十分な気道確保が、有意に高い合併症発生率と死亡率の原因と考えられている。
DM2患者で記載されている内分泌異常にはインスリン非感受性2型糖尿病および男性不妊の原因となる性腺不全がある。
DM1患者と同様DM2患者はIgGおよびIgMの両者の低下を伴う低ガンマグロブリン血症の頻度が高い。しかしながらそれに関連する臨床的な問題は報告されていない。
DM2患者で報告されている中枢神経系の異常には、MRIでみられる白質変化、PETでみられる前頭、側頭部の血流低下がある。このような解剖学的変化は認知機能、行動、人格に何らかの影響を及ぼすと思われるが、DM1と違いDM2では精神遅滞は認められない。
昼間の眠気、不眠症、レム行動障害およびレストレスレッグス症候群を含む様々な睡眠障害はDM2患者の症例報告や症例シリーズにおいて観察されている。
胃腸症状はDM2でしばしばあり、便秘、嚥下障害、腹痛などがある。
DM2の女性患者において、妊娠中、症状が悪化することがある。DM1の特徴とされている羊水過多はDM2患者では報告されていない。
発癌のリスク
いくつかの症例報告は、筋強直性ジストロフィー患者における癌発生率の増加を示唆している。最近の後ろ向き研究によると、DM2に罹患した患者は、全体的に癌を発症するリスクが高いようである。DM2患者の癌には、結腸、脳、甲状腺、膵臓、卵巣、および子宮内膜が関与し得る。
遺伝子型と表現型の関連
CCTGリピートの大きさと筋力低下の発症年齢や疾患の重症度の他の指標(白内障切除術の年齢など)との間に統計学的有意な相関関係は認められない。両方のCNBPアレルにCCTGリピート伸長を持つ患者の臨床型は、片方のアレルにCCTGリピート伸長を持つ兄弟や両親と重症度が変わらないことが報告されており、CCTGリピート数は臨床経過に影響しないと考えられる。
DM2患者のリピートの大きさとリピートが調べられた時の年齢の間には相関関係があり、リピート長が年齢に従い増加することが示唆される。
浸透率
疾患の浸透率は、患者が自らの症状をどの程度自覚できるかと医師が疾患の兆候を正確に特定し解釈できるかによって決まる。患者の家族とその担当医師はDM2の臨床的特徴をより認識できるようになるため、浸透率は100%近くになる。
経験のある医師による診察 経験のある複数の医師が18歳以上の成人234人を診察した研究によると、一人を除いてすべてのDM2患者を正確に診断した。(50歳男性の患者をおそらく不十分な診察の結果、正常であると分類した。)
家族歴によって以下のことが明らかになった。
- 経験のある医師によって診察されていなかった絶対保因者は、40歳代、50歳代では診断されなかったか、しばしば「リウマチ」、線維筋痛症、関節リウマチ、炎症性ミオパチー、非典型的運動ニューロン疾患、代謝性ミオパチーと誤って診断された。
- 若年発症の白内障、精巣機能不全、心臓不整脈などの骨格筋以外の症状がしばしば同定されたが、それがDM2の症状として認識されなかった。
表現促進現象
最初のDM2の特徴を決定する際に観察された家系において、世代を経るに従って症状が悪くなることが報告されている。このデータによると、症状の増悪は確認の際の先入観(不注意のためより若い世代の重症患者をその研究に含んでしまうこと)によるのではなく、表現促進現象によると考えられる。
- しかしながらDM1において表現促進現象の存在を確立した先天型のDM1に相当する先天型のDM2は存在しないことが、CNBP遺伝子の分子遺伝学的検査から明らかにされている。
- さらに疾患の重症度とCCTGリピート長が相関しないことは、世代間のリピート長の変化が必ずしも疾患の重症化をもたらすわけではないという結論を支持する。
- リピート伸長アレルをホモで持つ患者とヘテロで持つ兄弟は、臨床的に区別できないという観察によっても、リピート長と疾患の重症度との間に相関がないという事実は支持される。
学術名
国際筋強直性ジストロフィー協会(The International Myotonic Dystrophy Consortium (IDMC) とオンライン版ヒトメンデル遺伝 (Online Mendelian Inheritance in Man (OMIM))の両者はDM2と近位筋強直性ミオパチー(proximal myotonic myopathy (PROMM))は同一の疾患を指すとしている。PROMMの患者は初めDM1のいくつかの特徴を持つがDMPKの特異的三塩基リピートの伸長を持たないものとして記載された。PROMMとDM2の家系では臨床的な記載が異なることから、DM2とPROMMは当初臨床的に別の疾患と考えられていた。 しかしながらPROMMの家系のほとんどが現在ではDM2患者に特徴的なCNBP遺伝子の伸長を持つことが示されている。
注意:原因遺伝子変異が不明であり、こうした臨床型を示す場合にPROMMという用語は依然用いられることもある。しかしCNBPの分子遺伝学的検査で診断が確定した場合には、より正確なDM2という用語が推奨される。
多臓器を侵す筋強直性ジストロフィーのDM1,DM2以外の遺伝的原因の存在は指摘されているが、確立されていない。国際筋強直性ジストロフィー協会は新たに確立された多臓器を侵す筋強直性ジストロフィーに対して筋強直性ジストロフィーの新しい病型として順に番号を割り当てるとしている。
DM3であるとされた家系は後に家族性封入体筋炎を持ったパジェット病の珍しい表現型であったことが示された。この疾患はパジェット病と前頭側頭型認知症を伴った封入体筋炎としても知られ、VCP遺伝子の変異によっておこる。
頻度
筋強直性ジストロフィーは成人で最も多い筋ジストロフィーであり、全世界で約8000人に一人であると推定されている。DM1とDM2の割合は明らかではない。DM2の発症率は臨床的重症度に幅があるため不明のままである。
しかしながら、有病率はそれぞれの地域によって異なるとおもわれる。しかし、信頼のおける人口統計学上の研究はほとんどなされていない。DM2の有病率はドイツとポーランドで高く、ドイツ系、ポーランド系の人々で高いことが知られている。DM2はアフガニスタンやスリランカで報告されているが、中国、日本、サハラ以南のアフリカでは報告されていない。(訳注:日本では数家系が報告されている。)
遺伝学的に関連する(アレル)疾患
CNBP遺伝子の変異に関連した他の疾患は知られていない.
鑑別診断
多臓器を侵す筋強直性ミオパチー
現在明らかに筋強直性ジストロフィーの症状の原因となるのはDMPK遺伝子の3‘非翻訳領域にある翻訳されないCTGリピート伸長(筋強直性ジストロフィー1型、DM1)とCNBP遺伝子のイントロン1のCCTG伸長(DM2)のみである。この二型の筋強直性ジストロフィーの最終診断は分子遺伝学的検査を元に行われる。
通常の臨床評価で筋強直性ジストロフィーを特定することは可能であるが、真に成人発症である場合、DM1とDM2という二つの病型を臨床診断基準のみで信頼性を持って区別することは不可能である。DM1とDM2の患者の白内障を区別することはできない。DM1とDM2の最も大きな違いは内反尖足、新生児期の筋力低下および呼吸不全、精神発達遅滞、頭蓋・顔面異常、小児期の筋緊張低下および筋力低下がDM1患者では報告されているが、DM2患者では報告されていないことである。それ以外に、おそらくこれらの新生児期に受ける影響のため、DM1の成人患者はDM2の成人患者と比べしばしばより重症の筋力低下、筋強直現象を呈する; DM1患者では顔面および球筋の筋力低下、筋萎縮、心障害、中枢性過眠を含む中枢神経症状がより顕著である傾向がある。
家系内のある患者でCNBP CCTG伸長があることがわかり、DM2であることが確認された後、DM1およびDM2の遺伝子座と関連しない多臓器を侵す筋強直性ジストロフィー(すなわち非DM1,非DM2のPROMM症例)とした過去の報告は撤回されている。それでもなお、それ以外の遺伝的原因は存在するかもしれない。
新しい多臓器を侵す筋強直性ジストロフィー(DM3)を持つとされた家系はDM1、DM2に共通するいくつかの特徴(白内障、筋強直現象)を有していたが、まったく異なる神経系の異常(運動ニューロン病と海綿状脳症)を呈した。この家系の症状は当初筋強直性ジストロフィーと酷似していると考えられたが、現在ではVCPの変異によって起こるパジェット病を伴う家族性封入体筋炎によるもので、DMとは異なる疾患であったと考えられている。
遠位型ミオパチー
鑑別診断における他の主なグループは遠位型ミオパチーである(表2を参照)。
- Udd型遠位型ミオパチーは、足首の背屈の筋力低下と35歳以降かかとで歩くことができないことを特徴としている。疾患の進行は遅く、筋力低下は前脛骨筋に限定されている。10~20年後につま先の伸筋が障害されるようになり、歩行時に下垂足となり歩行がぎこちなくなる。Udd型遠位型ミオパチーは、タイチンをコードする遺伝子である TTNの突然変異によって引き起こされる。
- 埜中型早期成人型遠位型ミオパチーは縁取り空胞を伴い、通常、10~20歳に下肢の前面筋およびつま先の伸筋で症状が出始める。進行とともに下垂足や鶏歩が現れ、12~15年後には歩行能力の喪失に至る。これは、Quadriceps-sparing myopathyと同じ疾患であり、GNEの突然変異によって引き起こされる。
- Zaspopathy(Markesbery-Griggs型遅発性遠位型ミオパチー)(OMIM 609452)は、通常40代後半から始まる足背屈の筋力低下を特徴とし、その後、指や手首の伸筋と手の内在筋へゆっくりと進行する。最終的に、下肢の近位筋が障害される。この疾患は、LDB3(ZASPとしても知られる)の突然変異によって引き起こされる。
- MDP3は優性の遠位型ミオパチーであり、フィンランドの一家族で報告されている。上肢に初発症状が現れた人もいれば、下肢に発症した人もいる。後に、上肢と下肢の両方が障害される。
- 三好型早期成人型ミオパチーは、下肢の後面筋の症状で始まり、階段を上ったりつま先で歩くことが困難になり、LGMD2Bと同様に他の遠位筋および近位筋に進行する(ジスフェルリノパチー参照)。血清CK濃度は通常の50倍以上になる。
- Welander型遠位型ミオパチーは、通常の手指伸筋における発症ではなく、下肢の前面筋に症状が現れることがある。40歳以降に現れる人差し指の伸筋の筋力低下が典型的な症状であり、その後、他の指の伸筋、下肢の前面筋および後面筋へゆっくりと進行する。
表2 遠位型ミオパチー
| 障害 | 平均 初発年齢 |
初期に 障害される筋 |
血清 CK値 |
筋生検 | 遺伝子 (遺伝子座)1 |
|---|---|---|---|---|---|
| 常染色体優性 | |||||
| Welander型遠位型ミオパチー (OMIM 604454) |
>40歳 | 上肢の遠位筋(手指および手根伸筋) | 正常 あるいはやや上昇 |
縁取り空胞 | (2p13) |
| Udd型遠位型 ミオパチー |
>35歳 | 下肢の前面筋 | 縁取り空胞+/- | TTN | |
| Zaspopathy (Markesbery-Griggs型遅発性遠位型 ミオパチー) (OMIM 609452) |
>40歳 | 空胞性および筋原線維性 ミオパチー |
LDB3 (ZASPとしても 知られる) |
||
| 遠位型ミオチリン異常症 (OMIM 609200) | >40歳 | 下肢の後面筋>前面筋 | やや上昇 | 空胞および筋原線維 | MYOT |
| Laing型小児型 遠位型ミオパチー(childhood-onset distal myopathy, MPD1) |
<20歳 | 下肢の前面筋および 頸部屈筋 |
正常から(まれに)中等度 上昇 |
前脛骨筋のタイプ1線維の萎縮、近位筋における筋線維不均等 | MYH7 |
| 声帯および咽頭徴候を伴う遠位型 ミオパチー(Distal myopathy with vocal cord and pharyngeal signs , MPD2) |
35-60歳 | 左右非対称の下腿筋、手内筋、発声障害 | 1-8倍 | 縁取り空胞 | MATR3 |
| 凹足と腱反射消失を伴う遠位型 ミオパチー |
15-50歳 | 前面および後面の下腿筋、発声困障害、嚥下障害 | 2-6倍 | ジストロフィー変化、縁取り空胞 | (19p13) |
| 新規フィンランド型遠位型ミオパチー(New Finnish distal myopathy, MPD3) | >30歳 | 手内筋もしくは下腿前面筋 | 1-4倍 | ジストロフィー変化、縁取り空胞、好酸性封入体 | (8p22-q11, 12q13-q22) |
| 常染色体劣性 | |||||
| 埜中型早期成人型 遠位型ミオパチー |
15-20歳 | 下肢の前面筋 | <10倍 | 縁取り空胞 | GNE |
| 三好型早期成人型 ミオパチー |
下肢の後面筋 | >10倍 | 筋原性変化 | DYSF | |
- 遺伝子が不明な場合にのみ、遺伝子座を記載している。
筋強直現象 電気的ミオトニーは様々な状況で起こるが、家系の中の複数の例で筋強直現象の存在が認められる場合、診断はDMか非ジストロフィー性筋強直(クロライドおよびナトリウムチャンネル遺伝子の変異によって起こる先天性ミオトニー、先天性パラミオトニー、高カリウム性周期性四肢麻痺)に絞られる。これらはDM1やDM2の特徴である筋ジストロフィーとしての変化や多臓器疾患としての特性を示さず、そのために臨床的に区別することができる。
その他 DM2患者は非典型的運動ニューロン疾患、炎症性ミオパチー、線維筋痛症、関節リウマチ、代謝性ミオパチーと誤診されることがある。
臨床的マネジメント
最初の診断後の評価
筋強直性ジストロフィー2型(DM2)と診断された患者の症状の程度を明らかにするため、以下の内容について評価することが勧められる。
- 通常の診療での筋力および機能の評価
- DMの虹色後嚢下白内障に精通した眼科医による検査(初期状態をはっきりさせるため)
- 少なくとも以下の内容を含む初期の心機能評価
- 後の比較のため心電図記録
- 患者に症状がある場合や通常の心電図で明らかな不整脈や伝導障害がある場合のホルター心電図や侵襲的電気生理学的検査
- 心筋症のリスクがあるため、心エコー検査や心臓MRIの検討
- インスリン抵抗性や糖尿病の有無を評価するための空腹時の脂質分析、血糖およびHbA1c濃度を含むベースラインの血清学的検査
- 生殖機能を評価するための思春期以降の男性に対するテストステロンおよびFSH検査
- 甲状腺機能検査。甲状腺機能低下はDM2をおこす変異と関連するかははっきりしていないが、どのような原因であっても甲状腺機能低下症はDM2患者での筋力低下その他の症状の悪化に関連する。
- CK,トランスアミナーゼ(AST,ALT)、γGTP。AST,ALT,γGTPの異常は、肝細胞由来か筋由来かは不明であるが、それらの値はDM2患者でしばしば上昇する。基準となる異常トランスアミナーゼおよびγGTPの値を知ることで、不必要な肝臓の検査を避けることができる。
- DM2患者では、血清蛋白電気泳動と免疫電気泳動においてγ分画の減少が、IgGおよびIgMの低下により、しばしば認められるため、これらの値の個々の患者における基準を決めるための測定が必要となる。これらの変化の臨床的問題との関連はないが、DM2患者での異常な免疫グロブリンの値を知ることで個々の患者の基準を定め、将来低グロブリン血症を示す検査結果に対する誤った解釈を避けることができる。
臨床症状に対する治療
DM2に対する治療ガイドラインが発表されている。
リハビリテーション医、作業療法士、理学療法士は、病気の進行に伴い、短下肢装具、車いす、その他の補助器具の必要性の判断に役立つ。
通常の身体活動はDM2の筋力と持久力を保つ、あるいは骨格筋の疼痛管理に役立つと思われる。
筋強直現象は通常軽度であり、まれにしか治療を必要としない。しかしメキシレチンはある種の筋強直現象に対しては非常に有効であり、一部のDM2患者の疼痛管理に有用である。
個々の薬物及び薬物の組み合わせにより、疼痛管理に対する効果は様々である。一つの薬剤が一定して有効であることはない。ある程度有効であった薬剤としてはメキシレチン、ガバペンチン、非ステロイド性抗炎症薬(NSAIDS)、低用量甲状腺ホルモン補充、低用量ステロイド(例:5mgプレドニゾン隔日投与)、三環系抗うつ薬がある。麻薬性鎮痛薬は、包括的疼痛管理プログラムの一環として使われた場合有用であるかもしれないが、耐性が生じ、容量が増える可能性がある。
心臓の症状がある患者や心電図上不整脈の所見がある患者では、他の症状が出現する以前に致死性の不整脈が起こる可能性があるため、心臓専門医へのコンサルテーションが強く勧められる。心電図、ホルター心電図、心エコーは、失神、動悸その他の心臓由来の症状を評価するために行われるべきである。より進んだ方法として、侵襲的心臓電気生理学的検査が必要な場合がある。心筋症のリスクが高いため、心エコー検査と心臓MRIを考慮する必要がある。
明らかな不整脈があるDM2患者では除細動器の埋め込みが有効であるというエビデンスが増えてきているが、無症状の患者に対するペースメーカー、除細動器の役割については、はっきりとした結論は出ていない。
視力障害を起こす場合に、白内障を取り除くことができる。視力の変化は嚢下混濁の数だけではなく、その部位によって決まるため、典型的な高齢者の核性白内障と比べ、直接検眼鏡検査あるいは細隙灯検査を用いても、DM2患者での白内障の機能的重症度を過小評価することがある。
テストステロン補充療法は症状のある性腺機能不全に有効なことがある。
DM2での特徴的な胃腸症状は明確にはされていないが、一部の患者は食後の腹痛、腹部膨満、便秘、下痢などを訴える。筋強直性ジストロフィー1型でみられるように、一部の患者ではメトクロプロミド(商品名プリンペラン)、テガセロド(商品名ゼルノーム:訳注日本未発売)などの消化管運動促進薬が有効である。
二次的合併症の予防
DM2患者では麻酔のリスクが高まる可能性があるため、手術前後の心機能および呼吸機能の慎重な評価が推奨される。
DM2患者の筋力低下の進行は甲状腺機能低下と関連しているため、甲状腺機能低下症を治療することである程度の筋力が回復することがある。
経過観察
無症状のまま進行する心伝導障害および心筋症を検出するために年1回の心電図、心エコー検査できれば心臓MRIが推奨される。
施設によっては心症状がなくとも患者に年1回の24時間ホルター心電図を実施する。
年1回空腹時血糖、HbA1cを評価すべきである。
男性患者で、疲労感の増悪や性的活力の低下がある場合には性腺機能不全を検査するべきである。補充療法により効果がみられるかを判断するため、2-3年に一度は症状がなくても検査を行う必要がある。
避けるべき治療・状況
筋力低下の進行は、ある種の高脂血症治療薬の投与に関連するので、スタチン型高脂血症治療薬を中止すれば筋力が改善することがある。
注:DM2のすべての患者がスタチン製剤に副作用を示すわけではないので、DM2患者にこれらの薬剤を使うことは絶対的禁忌ではない。
リスクのある血縁者の評価
遺伝カウンセリング目的のリスクのある血縁者の検査に関する事項に関しては遺伝カウンセリングを参照。
研究中の治療法
広範囲の疾患に対する臨床研究の情報に調べる場合には米国のClinicalTrials.govあるいはヨーロッパのEU Clinical Trials Register を検索すること。
注:本疾患に対する臨床治験はされていない可能性がある。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
筋強直性ジストロフィー2型は優性遺伝形式で遺伝する。
患者家族のリスク
発端者の両親
- 現在の所、生物学的両親の分子遺伝学的検査が行われたすべての発端者は両親のどちらかがCNBP遺伝子の伸長を持つ。
- 新規突然変異を持つ発端者は報告されていない。
- CNBP遺伝子の伸長は世代間および体細胞内で不均一性を示す。一般に、リピートの長さは次の世代に伝わる際に短縮し、個人の中で年齢を経るにつれて伸長する(遺伝カウンセリングに関連した問題の体細胞内不安定性の項を参照)。母親由来、父親由来による伸長、短縮に関する一定の傾向はない。
発端者の同胞
- 発端者の同胞の危険性は両親の遺伝的状況による。
- 両親の片方が伸長したCNBP遺伝子の伸長を持つ場合は、各同胞は50%の可能性でその伸長を引き継ぐ。
発端者の子
- CNBP遺伝子の伸長を片方のアレルで持つ患者の子供は、それぞれ50%の確率で遺伝子の伸長を引き継ぐ。
- CNBP遺伝子のCCTGリピートは次の世代に遺伝する際、短縮し、個人の中で年齢を経るにつれて伸長する傾向にある。
発端者の他の家族
他の血縁者のリスクは、発端者の親の状況による。もし親も発症者であれば、その血縁者もリスクがある。
遺伝カウンセリングに関連した問題
新規病原性変異体が明らかな家族における考慮事項
常染色体優性疾患の発端者の両親がどちらも病原性変異体または障害の臨床的証拠を持っていない場合は、発端者が新規変異を持っている可能性がある。しかし、代理父や代理母(生殖補助医療など)または非公開の養子縁組などの非医学的説明も考えられる。
測定されたリピートの大きさと疾患の重症度との間に相関関係はない、そのため発症年齢や臨床経過を分子遺伝学的検査から推測することはできない。
体細胞内不安定性
CNBP遺伝子のCCTGリピート伸長は非常に不安定であり、年齢を経るに従い増加する傾向にある。異なる血液サンプルを使って複数回検査を行った場合、伸長の程度が異なることがある。さらに一回の末梢血サンプル中にも、サザンブロット解析で検出できる複数の伸長したアレルサイズが含まれている。一回のサンプルの中で主たるアレルの大きさからも、検出可能な異なる伸長の合計からも疾患の重症度、発症年齢、臨床症状を予測できない。
無症状のリスクのある成人の検査
無症状のリスクのある成人の検査は、分子遺伝学的検査の項で記述した方法により実施可能である。予測検査を行うことによって、被検者がCNBP遺伝子の伸長を持っているかすなわち被検者が発病するリスクがあるかどうかを判定することができる。リピートの大きさから、発症年齢、重症度、臨床症状を予測することはできない。リスクのある家系の人に検査の提案をする前に、症状のある人の遺伝子検査をして、その家系で本当にCNBP遺伝子の伸長があるかを確認する必要がある。分子遺伝学的検査の限界と予測検査に関連するリスクを理解するため、それぞれの被検者は、予測検査の前に遺伝カウンセリングを受けるべきである。
無症状のリスクのある18歳未満の未成年の検査
本疾患は成人発症であり、病気を阻止したり、転帰を改善させたりする有効な治療法がないことから、無症状のリスクのある18歳未満の人の検査は勧められない。症状のある18歳未満の人に対して遺伝子検査をすることは、診断を確立するという利点がある。the National Society of Genetic Counselors position statement on genetic testing of minors for adult-onset conditions やthe American Academy of Pediatrics and American College of Medical Genetics and Genomics policy statement: ethical and policy issues in genetic testing and screening of childrenを参照のこと
家族計画
- 遺伝学的リスク評価や出生前検査の可否などについての議論は妊娠前に行うのが望ましい。
- 罹患しているあるいはリスクのある若者に対する遺伝カウンセリング(子孫や出産の選択に対する潜在的リスクに関する議論を含む)の提案を行うのが望ましい。
DNAバンク DNAバンクは主に白血球から調製したDNAを将来利用することを想定して保存しておくものである。検査技術や遺伝子、変異、あるいは疾患に対するわれわれの理解が将来さらに進歩すると考えられるので、DNA保存が考慮される。
出生前検査と着床前遺伝子診断
罹患している家族においてCNBP遺伝子伸長が確認されると、リスクのある妊娠に対する出生前診断と着床前遺伝子診断が可能である。
特に初期の診断というより中絶の目的で検査を検討している場合、出生前診断の実施に関して医療従事者と家族の間で視点が異なる場合がある。多くの医療施設では、両親が出生前診断を受けるかどうかを選択するとしているが、これらの問題に対する十分な議論を行うことが望まれる。
(訳注:本邦では着床前診断に対応可能な施設はきわめて少なく、個別の倫理審査が必要など利用には障害が多い。)
更新履歴:
-
Gene Reviews著者: Robert Pilarski, MS, LGC, MSW, Karan Rai, BS, Colleen Cebulla, MD, PhD and Mohamed Abdel-Rahman, MD, PhD
日本語訳者: 朝倉 一 ,古屋充子(横浜市立大学医学研究科分子病理)
GeneReviews 最終更新日:2016.10.13 日本語訳最終更新日: 2019.3.22. - Gene Reviews著者: Joline C Dalton, MS; Laura PW Ranum, PhD; John W Day, MD, PhD 日本語訳者: 堀江 里歩、(監訳) 高橋 正紀(大阪大学大学院医学系研究科保健学専攻 機能診断科学講座) Gene Reviews 最終更新日:2013.7. 3. 日本語訳最終更新日: 2020.01.05. (in present)