難聴・遺伝性難聴概説
(Deafness and Hereditary Hearing Loss Overview )
Gene Review著者: Richard JH Smith, MD, Guy Van Camp, PhD
日本語訳者: 窪田美穂(ボランティア翻訳者),岸本 洋子(島田療育センターはちおうじ)
Gene Review 最終更新日: 2014.1.9. 日本語訳最終更新日: 2015.3.25
原文 Deafness and Hereditary Hearing Loss Overview
要約
疾患の特徴
伝性難聴は,伝音難聴もしくは感音難聴,またはその両方である.また,症候性(外耳やその他の器官の奇形や,その他の臓器系の疾患を伴う場合),もしくは非症候性(外耳の目にみえる奇形や関連疾患がない場合)である.言語習得前難聴(言語発達以前)の場合も言語習得後難聴(言語発達以降)の場合もある.
診断・検査
遺伝性難聴は後天性(非遺伝性)の難聴と鑑別されなければならない.遺伝性難聴は耳科的,聴覚的,身体的検査や家族歴,補助的検査(例えば,側頭骨のCT画像診断),分子遺伝学的検査により診断される.分子遺伝学的検査は臨床施設で症候性・非症候性難聴の多くの病型に対して行われており,診断と遺伝カウンセリングの場でとりわけ重要な役割を果たしている.
臨床的マネジメント
症状の治療:
遺伝性難聴は,耳鼻咽喉科医,聴覚士,臨床遺伝医,小児科医によるチーム医療を行う.難聴者教育の専門家,神経科医,小児眼科医が含まれることもある.治療には聴覚補助器具や振動補聴器といった適切な補助医療機器の選択が含まれる.生後12カ月以降の重度~最重度の難聴児に対しては,人工内耳埋込術が考慮される.補聴器,耳科手術,人工内耳埋込術による早期の聴覚的介入が言語習得前難聴児の最適な認知力発達にとっては不可欠である.
経過観察:
聴覚検査を継続して行い,難聴の安定もしくは進行を評価する.中耳浸出液などの別の難聴要因がみられる場合には治療すること.
回避すべき薬剤と環境:
騒音への曝露.
リスクのある近親の検査:
遺伝性難聴のリスクのある小児に対しては,分子遺伝学的検査 (家系特異的変異が分かっている場合)と スクリーニング目的の聴力検査を実施すべきである.
遺伝カウンセリング
遺伝性難聴は常染色体優性, 常染色体劣性, X連鎖性劣性,あるいはミトコンドリア遺伝の形式をとる.遺伝カウンセリングとリスク評価には各症例に対する正確な遺伝学的診断が重要である.具体的な診断がつかない場合には,GJB2遺伝子やGJB6遺伝子の分子遺伝学的検査結果を加味した経験的再発率を遺伝カウンセリングに用いる.
診断
臨床症状
難聴は病型と発症時期により分類される:
病型
- 外耳や中耳小骨の異常による伝音難聴.
- 内耳構造(すなわち,蝸牛)の機能障害による感音難聴.
- 混合性難聴とは伝音難聴と感音難聴がどちらもみられる場合である.
- 中枢性聴覚障害は第8脳神経,聴性脳幹,大脳皮質における損傷もしくは機能不全により起こる.
発症時期
- 言語習得前難聴は言語発達以前に難聴がみられる場合である.先天性(出生時にみられる)難聴はすべて言語習得前難聴であるが,言語習得前難聴のすべてが先天性ではない.
- 言語習得後難聴は正常な発話言語発達の後に起こるものである.
難聴の重症度. 聴覚単位はデシベル(dB)で計測される.周波数ごとに掲げられた閾値または0dBは正常な若年成人が50%の確率でトーンバーストを感知する水準を示している.正常範囲の15db以内の閾値の場合,聴覚正常であると判断される.難聴の重症度を表1に示す:
表1. 難聴の重症度(デシベルdB)
| 重症度 | 聴覚域(デシベルdB) |
|---|---|
| 軽度 | 26~40 dB |
| 中等度 | 41~55 dB |
| 準高度 | 56~70 dB |
| 高度 | 71~90 dB |
| 重度 | 90 dB |
注: 1)会話での発話は約50~60 dB HL(聴覚レベル)であるため,機能的障害を純音平均に基づいて算出すると間違いやすい.例えば,45dBの難聴は機能的には30%の難聴よりもずっと重篤である.2)聴覚の制限が言語発達へ与える影響が著しいと考えられる年少児に対しては,これ以外の評価方法を用いる方が良い. [Northern & Downs 2002].
表2.難聴パーセント
| 難聴% | 純音平均(dB) 1 | 残存聴覚% |
|---|---|---|
| 100% | 91 dB | 0% |
| 80% | 78 dB | 20% |
| 60% | 65 dB | 40% |
| 30% | 45 dB | 70% |
- 500 Hz,1000 Hz,2000 Hz,3000 Hzの純音平均
周波数ごとの難聴. 難聴における周波数は以下のように定義される:
- 低周波(500 Hz未満)
- 中周波(501~2000 Hz)
- 高周波 (2000 Hz以上)
「聴覚障害hearing impairment」と「難聴hearing loss」とは,どちらも医療専門家により同義語として用いられることが多く,聴力測定で正常範囲以下の聴力を指す.
deaf("d"は小文字)
聴力測定で聴覚範囲が高度~重度の場合を指す口語表現である.
Deaf culture ("D"は常に大文字)
米国のDeafコミュニティのメンバーはdeafであり,使用言語はAmerican Sign Languageである.他の文化と同様,メンバーは独特の社会的・社交的属性を持つ. Deafコミュニティ(the Deaf)のメンバーは自らの聴覚が「障害を受けている」とは考えておらず,聴覚を「喪失した」とも考えていない.むしろ,自覚的にdeafである.彼らのdeafnessは治療や治癒の対象となる病態でも疾患でもない.
聞こえの悪さ.
この言葉は聴覚学的よりも機能的な意味あいをもつ.the Deafでは聴力が幾分ある場合を指して用いられているが,軽度~重度の聴覚喪失まで様々である.Deafコミュニティではdeafの人々は発話言語を用いないが,耳の聞こえが悪い人はいくらか発話言語を用いる.
確定診断
生理学的検査
生理学的検査では聴覚系の機能状態が客観的に測定でき,どの年齢でも実施可能である.生理学的検査には以下が含まれる:
- 聴性脳幹反応検査 (ABR, BAER, BSER) 聴性脳幹反応検査では電気生理学的反応を誘発する刺激(クリック音)が用いられる.この反応は第8脳神経および聴性脳幹で発生するものであり,頭皮上の電極で記録される.腸性脳幹反応検査(ABR)の「V波検出閾値」は神経学的に正常な者の1500~4000Hzの聴覚感受性に最も一致する.聴性脳幹反応検査(ABR)では低周波(1500Hz未満)の感受性は評価できない.
- 聴性定常反応聴力検査(ASSR) 聴性定常反応聴力検査はオーストラリア,アジア,カナダでのみ用いられていた聴力の電気生理学的測定法であるが,現在では米国や欧州での使用が増えている.聴覚反応が持続的な音刺激の変化に対応する時期がみられるかを,皮膚電極から測定する.刺激が継続したシグナルであるために,到達されるまでの平均音圧レベルはクリック音を用いる聴性脳幹反応検査(ABR)の値よりも高い.この違いにより,聴性定常反応聴力検査(ASSR)では聴性脳幹反応検査(ABR)で反応を示さない小児の聴覚感受性を測定できる場合が多い.
- 誘発耳音響放射 (EOAEs) 誘発耳音響放射では,蝸牛における発生音をマイクロフォン変換器のついたプローブを用いて外耳道で測定する.誘発耳音響放射(EOAE)は主に広範囲の周波数域に及ぶ蝸牛の外有毛細胞の活動を示す.40-50dB聴覚レベルより良好な聴覚感受性を持つ耳に用いられる.
- イミタンス検査は中耳圧,鼓膜可動性,耳管機能,中耳小骨の可動性といった末梢聴覚系を評価する.
- イミタンス検査(ティンパノメトリー検査,音響反射閾値,音響反射消失)
聴力検査
聴力検査では人がどのように聴覚情報を処理しているか,すなわち聞いているのかを,主観的に調べる.聴覚検査では聴性行動反応検査と純音聴覚検査が行われる.
- 聴性行動反応検査には,聴性行動反応聴力検査(BOA)と視覚増強聴力検査(VRA)がある.BOAは生後直後から6か月の乳児に用いられるが,試験を行う側の技能に大きく左右されやすく,誤りが生じやすい.VRAは生後6か月から2.5歳の小児に用いられるものであり,信頼性の高い完全な聴力図が得られるが,小児の成熟年齢と検査を行う側の技能に左右される.
- 純音聴覚検査(気導および骨導) 純音聴覚検査では人が純音を周波数の関数(ピッチ)として「聞く」最低の強さを調べる.250(まんなかのC音に近い音)から8000Hzまでのオクターブ周波数を,イヤホンを用いて検査する.強さ,すなわち大きさはデシベル(dB)で測定され,2つの音圧間の比と定義される.0dB聴力レベルは成人の正常聴覚の平均閾値である.120dB聴力レベルは痛みを引き起こすほどの大きさである.語音了解閾値レベル(SRT)や語音明瞭度が評価される.
- 気導聴力検査 気導聴力検査ではイヤホンを通じた音を聞く.閾値は外耳道,中耳,内耳の状態による.
- 骨導聴力検査 骨導聴力検査では乳様突起もしくは頭頂骨に設置することにより外耳と中耳の影響を除いた音を,振動器を通じて聞く.閾値は内耳の状態による.
- 条件遊戯聴力検査(Conditioned play audiometry;CPA) 条件遊戯聴力検査は2.5歳から5歳の小児に対して行われる.協力的な子どもであれば,左右の耳に関して周波数特異的な完全な聴力図が得られる.
- 従来の聴力検査 5歳以上に対しては,従来の聴力検査が行われる.音が聞こえたら知らせるというやり方である.
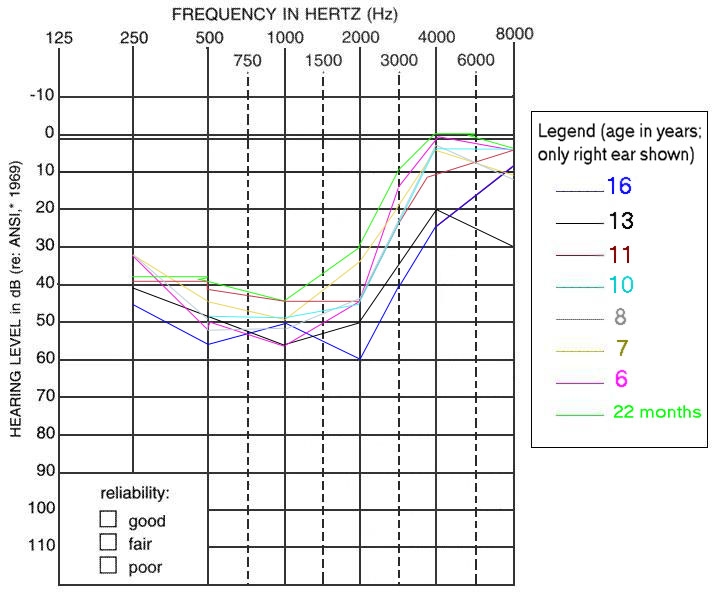
Figure 1 (link of GeneReviews)
図1. 聴力プロファイル
その他
- 先天性難聴は新生児に対する聴覚スクリーニング検査(NBHS)によりみつかる.NBHSは米国国立衛生研究所(NIH)が推進している.新生児に対する聴覚スクリーニング検査は米国の33の州とコロンビア特別区では実施が法律もしくは規則で義務付けられている.残りの州のほとんどでも新生児に対する聴覚スクリーニング検査が広く実施されているが,まだ義務付けられてはいない.カリフォルニア州とテキサス州は例外で,新生児に対する聴覚スクリーニング検査が行われるのは特定の州民もしくは要請のあった場合のみである [National Newborn Screening and Genetics Resource Center, National Newborn Screening Status Report (pdf)].
- 親が難聴の可能性や言語発達の遅れを心配している場合には,どの小児に対しても聴覚スクリーニング検査を行う必要がある.
鑑別診断
言語発達が遅れている小児に対しては,聴覚系への評価を行うこと.聴力検査が正常で進行性の言語消失や側頭葉性てんかんがみられる場合には, Landau-Kleffner症候群を考慮すること.
自閉症の年少児では,言語発達遅滞により難聴が疑われることもある 「自閉症概説を参照のこと」).
頻度
難聴は最も多い先天性異常であり,先進国では最もよくみられる感覚神経障害である [Hilgert et al 2009].新生児500人中1人が両側性感音難聴(40dB以上)であり,青年期までには頻度は1000人中3.5人に上昇する[Morton & Nance 2006].
症候性難聴と常染色体優性の非症候性難聴が言語習得前難聴の中で占める割合は小さい.言語習得前難聴の50%以上が遺伝性であり,常染色体劣性の非症候性難聴が最も多い.常染色体劣性の非症候性難聴の約50%は,(コネキシン26蛋白をコードする)GJB2 遺伝子と(コネキシン30蛋白をコードする)GJB6遺伝子の変異によって生じるDFNB1型難聴である.常染色体劣性の難聴の原因となるGJB2遺伝子変異の保因者頻度はおよそ33人に1人である.
一般人口集団では難聴の頻度は年齢とともに増す.この変化は遺伝要因および環境要因を反映するだけでなく,環境誘発要因と個人の遺伝的易罹患性との相互関係も反映している.これは,アミノグリコシド誘発性の中毒性難聴(「非症候性難聴およびミトコンドリア性難聴」の項を参照のこと),中耳浸出や,おそらく耳硬化症にもあてはまる.
原因
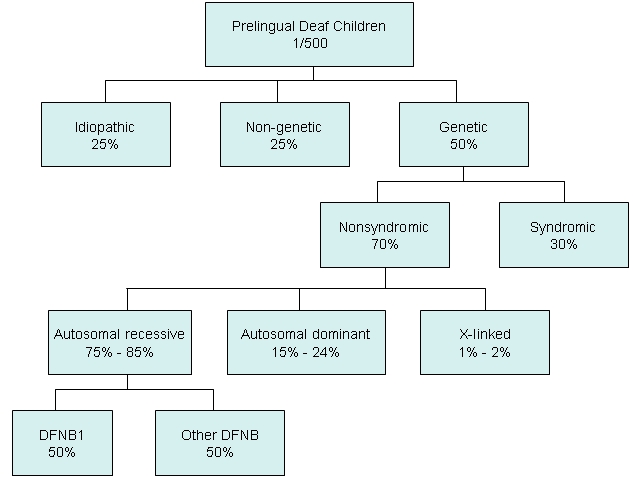
Gi
lFigure 2 (ink of GeneReviews)
図2. 小児における言語習得前難聴(40dB以上)の原因
環境要因
小児の後天性難聴 小児の後天性難聴は,一般に「TORCH」と呼ばれる微生物(すなわち,トキソプラズマ,風疹ウイルス,サイトメガロウイルス,ヘルペスウイルス)の出生前感染や,出生後の感染,とりわけ髄膜炎菌,インフルエンザ菌,肺炎球菌による細菌性髄膜炎によって起こる.大腸菌,Listeria monocytogenes,Streptococcus agalactiae,Enterobacter cloacae といった他の微生物による髄膜炎でも難聴となることがある.
しかし,先進国では先天性難聴の環境的非遺伝的原因のうち最も多いのは先天性サイトメガロウイルス(CMV)感染症である.全出産数に対する頻度は約0.64%であり,このうちの10%が症候性サイトメガロウイルス感染症である.症状がみられない場合には,小学校入学前に最大4.4%に片側性もしくは両側性難聴がみられるが,これには著しい民族間のばらつきがある.サイトメガロウイルス感染症による難聴の診断は困難であり見過ごされる場合が多く,症状の変わりやすい様々な感音難聴とされることがある[Kenneson & Cannon 2007].
成人の後天性難聴 成人の後天性難聴は環境要因によるものが最多であるが,難聴になりやすいかどうかは環境-遺伝的相互関係に最も影響される.年齢に相関した難聴や騒音性難聴は複雑な「環境-遺伝的」難聴を示す例としては最多のものだ.しかし,現在のところ,これらの複雑な難聴と関連づけられている遺伝子はわずかである[Huyghe et al 2008, Konings et al 2009].
アミノグリコシド誘発性難聴がミトコンドリアDNAの1555番塩基でAからGへの置換がみられる人に起こりやすいという所見は,医療との関連のある環境要因を示す1例である(「非症候性難聴およびミトコンドリア性難聴」の項を参照のこと).
遺伝要因
単一遺伝子疾患
症候性難聴
症候性難聴は外耳やその他の器官の奇形,もしくは他臓器疾患を伴う難聴である.
非症候性難聴
非症候性難聴は外耳の目に見えるような異常やその他の疾患を伴わない.しかし,中耳や内耳の異常を伴っている可能性がある.
この概説ではよくみられる症候性および非症候性難聴の臨床的特徴と分子遺伝学に焦点を当てている.GeneReviewsに掲載されている疾患についてはリンク先を参照のこと.
症候性難聴
難聴を伴う400以上の遺伝性症候群が報告されている[Toriello et al 2004].症候性難聴は言語習得前難聴の最大30%を占めるとも言われているが,言語習得後難聴の発症率と診断を反映して,難聴者全体ではその相対的割合はずっと低い.ここでは症候性難聴を遺伝形式ごとに論じる
常染色体優性の症候性難聴 .
Waardenburg症候群 (WS) Waardenburg症候群は常染色体優性の症候性難聴で最も多いものである.様々な程度の感音難聴と皮膚,髪(前頭部白髪),目(虹彩異色症)の色素異常が特徴である.患者が髪を染めることもあるために,前頭部白髪については病歴や身体診察により評価すること.その他の異常の有無に基づき,WS I型,WS II型,WS III型,WS IV型の 4つの亜型に分類されている.WS I型とWS II型は多くの共通点を持つが,大きな表現型の違いが1つある.すなわち,WS I型では両側内眼角の外側変位がみられるがWS II型ではみられない.WS III型では上肢に異常がみられる.WS IV型ではHirschsprung病を伴う.WS I型とWS III型はPAX3遺伝子変異が原因である.WS II型の一部の症例はMITF遺伝子変異による.WS IV 型はEDNRB遺伝子変異,EDN3遺伝子変異,SOX10遺伝子変異により発症する.
鰓弓耳腎症候群 (BOR) 鰓弓耳腎症候群は常染色体優性の症候性難聴のうち2番目に多くみられる.伝音難聴,感音難聴,混合型難聴に加えて,鰓弓嚢胞・瘻,耳介前孔を含めた外耳奇形,腎異常を伴う.浸透率は高いが,発現率は極めて多様である.BORの表現型が認められる家系の約40%でEYA1遺伝子変異が確認される.その他の家系のうち少数でSIX1遺伝子[Ruf et al 2004]およびSIX5遺伝子[Hoskins et al 2007]の変異が見つかっている.他のまだ同定されていない遺伝子によってもBORは発症する.
Stickler症候群 Stickler症候群は,進行性の感音難聴,口蓋裂,骨関節炎となる脊椎骨端異形成症が特徴である.分子遺伝学的異常に基づき,STL1 (COL2A1)型,STL2 (COL11A1)型,STL3 (COL11A2)型の3つのタイプが確認されている.STL1型とSTL2型では網膜剥離に至る重症の近視が特徴的である.COL11A2遺伝子が眼では発現しないところから,この所見はSTL3型にはみられない.
神経線維腫症2型 (NF2) 神経線維腫症2型はまれな,治療可能性がある難聴を伴う.NF2は両側性前庭神経鞘腫に続発する難聴であることが大きな特徴である.難聴が始まるのは通常20歳代であり,前庭神経鞘腫の腫大に併発する.一般に,片側性で進行は緩徐であるが,両側性で突発性の場合もある.後迷路性障害が聴覚検査で診断されることが多いが,確定診断にはガドリニウム造影剤を用いたMRI画像診断が必要である.患者には,髄膜腫,星状細胞腫,上衣細胞腫,髄膜血管腫症といった他の様々な腫瘍の発症リスクがある.NF2遺伝子変異が原因である.症状が発現していないリスクのある家系近親には分子遺伝学的検査により早期診断と早期治療が可能となる.
常染色体劣性の症候性難聴
Usher症候群
Usher症候群は常染色体劣性の症候性難聴のうちで最も多い.Usher症候群では2つの感覚障害がみられる.患者は生まれつき感音難聴を有するが,その後網膜色素変性症(RP)を発症する.米国の難聴盲目者の50%以上がUsher症候群である.網膜色素変性症による視覚障害は通常10歳までは現れないため,10歳前の眼底検査は有用性が限られている.しかし,網膜電図(ERG)で2歳から4歳の年少児の光受容体機能異常を確認することできる.10歳代では夜盲症と周辺視の喪失が現れ始め,視覚障害は着実に進行する.
難聴の程度と前庭機能検査の結果に基づき,Usher症候群は3タイプに分類されている.
- Usher症候群1型 先天性の重度~最重度の感音難聴と前庭機能異常が特徴である.患者は従来の補聴器では効果がみられず,通常コミュニケーションは手話である.前庭機能障害により,お座りや歩行といった運動発達は正常よりも遅れる場合が多い.
- Usher症候群2型 先天性の軽度~重度の感音難聴と正常な前庭機能が特徴である.聴覚補助具による増幅が有効であり,通常コミュニケーションは話し言葉である.
- Usher症候群3型 進行性の難聴と進行性の前庭機能障害が特徴である.
Pendred症候群は常染色体劣性の症候性難聴のうち2番目に多いタイプである.Pendred症候群の特徴は,通常(ばらつきはあるが)重度~最重度の先天性の感音難聴と,甲状腺機能正常の甲状腺腫である.出生時に甲状腺腫はみられないが,思春期初期(40%)もしくは成人期(60%)に発症する.甲状腺によるヨウ素の有機化の遅延は過塩素酸(perchlorate)排出試験によって確認できる.難聴は,側頭骨のCT検査により診断される骨迷路異常(Mondini型異形成もしくは前庭水管拡大症)に起因する.患者の大多数で前庭機能の異常がみられる.家系の約50%でSLC26A4遺伝子変異が同定されている.Mondini型異形成もしくは前庭水管拡大症と,進行性難聴がみられる患者には,このような遺伝学的検査を行うとよい.
初期の研究では,Pendred症候群は先天性難聴の最大7.5%を占めると報告されていたが,最近の報告ではペンドレッド 症候群の割合は低くなっている.SLC26A4遺伝子変異はDFNB4型非症候性難聴の発症原因でもある.
Jervell and Lange-Nielsen症候群 Jervell and Lange-Nielsen症候群は常染色体劣性の症候性難聴で3番目に多いものである.Jervell and Lange-Nielsen症候群の特徴は,先天性難聴と心電図で検出されるQT間隔延長(補正QT間隔の異常>440 m/秒)である. 患者には失神発作の既往があり,突然死にいたることもある.スクリーニングの心エコー検査の感受性は高くないが,難聴小児を対象としたスクリーニング検査を行うのが適切だろう.リスクの高い小児(すなわち,突然死,乳幼児突然死症候群(SIDS),失神発作,QT延長症候群の家族歴が陽性である場合)には,徹底した心機能評価を行うこと.患者には2つの遺伝子に変異がみられる.
ビオチニダーゼ欠損症 ビオチニダーゼ欠損症は4つのカルボキシラーゼと共有結合する水溶性ビタミンB群であるビオチンの欠損により発症する.ビオチンが共有結合するカルボキシラーゼは,糖新生に不可欠なピルビン酸カルボキシラーゼ,脂肪酸合成に不可欠なアセチルCoAカルボキシラーゼ,幾つかの分枝鎖アミノ酸の異化作用に不可欠なプロピニルCoAカルボキシラーゼとβメチルクロトニールCoAカルボキシラーゼである.ビオチニダーゼ欠損症が看過され,ビオチンの追加摂取により日常の食事が補正されない場合,患者には視覚障害に加えて痙攣,筋緊張亢進,発達遅滞,運動失調といった神経学的所見が現れる.ある程度の感音難聴が有症状小児の75%以上にみられる. 皮膚症状も起こり,発疹,脱毛症,伝染性結膜炎が起こる.ビオチン治療により神経症状や皮膚症状は解消するが,難聴と視神経萎縮は治らないのが普通である.従って,小児に発作性・進行性の運動失調と進行性感音難聴がみられる場合には,神経症状や皮膚症状の有無にかかわらず,必ずビオチン欠損症を考慮すること.代謝性昏睡を予防するために,できるだけ早急に食事治療と治療を開始すること[Heller et al 2002, Wolf et al 2002].
Refsum病 Refsum病は重度の進行性感音難聴と網膜色素変性症が特徴であり,フィタン酸代謝異常が原因である.極めて稀な疾患であるが,食事療法と血漿交換療法で治療可能であるため,難聴者に対してRefsum病を考慮することは重要である.血清フィタン酸値の測定により診断可能である(「ペルオキシソーム形性異常症・ツェルウェーガー症候群スペクトラム」を参照のこと).
X連鎖性症候性難聴
Alport症候群 Alport症候群の特徴は,様々な重症度の進行性感音難聴,末期腎不全に至る進行性糸球体腎炎,様々な眼科所見(例えば,前部円錐水晶体)である.通常,難聴は10歳以前にはみられない.常染色体優性,常染色体劣性,X連鎖性が報告されている.X連鎖性が症例の約85%であり,常染色体劣性は約15%,常染色体優性の報告も時々みられる.
Mohr-Tranebjaerg症候群 (難聴-ジストニア-視神経萎縮症候群) Mohr-Tranebjaergは,進行性の言語習得後非症候性難聴症候群を認めるノルウェーの大家系で最初に報告された.この家系を再調査したところ,視覚障害,ジストニア,骨折,精神遅滞といった追加所見が得られた.このことから,この難聴は非症候性ではなく症候性であることが示された.この症候群の原因遺伝子であるTIMM8A遺伝子は,細胞質ゾルからミトコンドリア内膜へ(TIM複合体),そしてミトコンドリア・マトリックスへの蛋白質の膜輸送に関与している.
ミトコンドリア性症状性難聴
ミトコンドリアDNA変異 ミトコンドリアDNAの変異により様々な疾患が起こる.このなかには,Kearns-Sayre症候群(「ミトコンドリアDNA欠失症候群」を参照のこと),MELAS, MERRF,NARPといった稀な神経筋疾患から,糖尿病,Parkinson病,Alzheimer病といったよくみられる疾患にまで多岐にわたる (「ミトコンドリア異常症概説」を参照のこと).日本では糖尿病患者の2~6%にMTTL1遺伝子の3243 A-to-G転位という変異が見つかっている.糖尿病患者でこの変異のある者の61%が難聴である.難聴は感音性であり,糖尿病の発症後に進行する.同じ変異がMELASにも関わっており,浸透率と組織特異性に関する疑問が浮上してくるが, ヘテロプラスミーの問題もあり複雑である.
非症候性難聴
遺伝性難聴の70%以上は非症候性である[Van Camp et al 1997].この項は遺伝形式ごとに論じる.
様々な遺伝子座が非症候性難聴(DFN)に関わっている.遺伝子座は遺伝形式に基づいて命名されている:
- DFNA: 常染色体優性
- DFNB: 常染色体劣性
- DFNX: X連鎖性
上記の名称の後に添えられる数字は遺伝子のマッピング・発見された順番を表している.
言語習得前非症候性難聴では,75~80%が常染色体劣性,20~25%が常染色体優性,1~1.5%がX連鎖性である.言語習得後非症候性難聴には同様のデータはないが,報告されている家系のほとんどが常染色体優性である.
常染色体劣性と常染色体優性 :
- DFNB1とDFNA3;どちらも13q12にマッピングされており,GJB2遺伝子とGJB6遺伝子の変異が原因である.
- DFNB2とDFNA11;どちらも11q13.5にマッピングされており,MYO7A遺伝子変異が原因である.この遺伝子変異はUsher症候群1B型の原因でもある.
- DFNB21とDFNA8/12;どちらもTECTA遺伝子変異が原因である.
非症候性および症候性の両方を認めるものには以下が含まれる:
- DFNB18とUsher症候群1C; USH1C遺伝子変異が原因である.
- DFNB12とUsher症候群1D; CDH23遺伝子変異が原因である.
- DFNB4とPendred症候群;SLC26A4遺伝子変異が原因である.
- DFNA6/14/38 とWolfram症候群;WFS1遺伝子変異が原因である.
大多数の常染色体劣性の遺伝子座は言語習得前の高度~重度の難聴の原因となる.例外はDFNB8であり,言語習得後に急速に進行する.
大多数の常染色体優性の遺伝子座は言語習得後難聴の原因となる.
- DFNA3,DFNA8,DFNA12,DFNA19といったいくつかの例外がある.
- DFNA6/14/38は主に低周波難聴を引き起こすものとしても注目される.
X連鎖性の非症候性難聴は言語習得前の場合もあれば言語習得後の場合もある.DFN3は混合型難聴である.
遺伝子型-表現型の相関関係が幾つか確認されている.例えば, TECTA遺伝子によりコードされる蛋白であるαテクトリンは3つの異なるドメインを持つ.すなわち,エンタクチンG1(ENTG1)ドメイン,フォン・ウィルブランド因子のタイプDリピート0-4 (VWFD 0-4)を持つゾナドヘシン(ZA)ドメイン,透明帯(ZP)ドメインである.
- DFNA8/12では,TECTA遺伝子変異はミスセンス変異であり,聴力プロファイルは変異の位置によって決まる.ZPドメインのミスセンス変異は安定性もしくは進行性の中周波難聴の原因となる. しかし,ZAドメインのミスセンス変異の場合,進行性の高周波難聴となる.
- DFNB21では, 変異により未熟な短縮蛋白が生じ,ヌル対立遺伝子のように振舞う.この中にはフレームシフト変異,ナンセンス変異,欠失がある.全ての場合で言語習得前の,左右対称的な難聴が起こり,重症度は中等度~重度である.
常染色体優性の非症候性難聴
常染色体優性の非症候性難聴の家系研究では,常染色体劣性の非症候性難聴の場合と同様に,異質性が高いことが示された.しかし,世界の多くの民族で常染色体劣性の非症候性難聴が単一の遺伝子(GJB2遺伝子)の変異によって生じているのに対し,常染色体優性の非症候性難聴では,大多数の症例の原因遺伝子は一つも同定されていない.
聴力プロファイル測定では,原因が分かっている常染色体優性の非症候性難聴者の1年間あたりの難聴の発症率を予測する際に用いることができる[Hildebrand et al, 2008].
さらに,常染色体優性の非症候性難聴における聴力プロファイルは独自の特徴を示すために(表3を参照のこと),分子遺伝学的検査の評価手順を決定する際に役立つ[Hildebrand et al, 2008](「評価手順」を参照のこと)
注:非症候性難聴の多遺伝子検査は検査手順および遺伝学的解析で述べる.
表3. 常染色体優性の非症候性難聴の原因となる既知の遺伝子に関する臨床症状と分子遺伝学
| 遺伝子座 | 遺伝子 | 発症時期 | 聴力プロファイル |
|---|---|---|---|
| DFNA1 | DIAPH1 | 言語習得後・10歳未満 | 低周波・進行性 |
| DFNA2 | KCNQ4 | 言語習得後・10歳代 | 高周波・進行性 |
| DFNA2B | GJB3 | 言語習得後。30歳代 | 高周波・進行性 |
| DFNA3 | GJB2 | 言語習得前 | 高周波・進行性 |
| GJB6 | |||
| DFNA4 | MYH14 | 言語習得後 | 均一・緩徐に右下がり |
| DFNA5 | DFNA5 | 言語習得後・10歳未満 | 高周波・進行性 |
| DFNA6/14/38 | WFS1 | 言語習得前 | 低周波・進行性 |
| DFNA8/12 | TECTA | 中周波・難聴 | |
| DFNA9 | COCH | 言語習得後・10歳代 | 高周波・進行性 |
| DFNA10 | EYA4 | 言語習得後・20-30歳代 | 均一・緩徐に右下がり |
| DFNA11 | MYO7A | 言語習得後・10歳未満 | |
| DFNA13 | COL11A2 | 言語習得後・10歳代 | 中周波 |
| DFNA15 | POU4F3 | 言語習得後 | 高周波・進行性 |
| DFNA17 | MYH9 | 言語習得後 | 高周波・進行性 |
| DFNA20/26 | ACTG1 | 言語習得後 | 高周波・進行性 |
| DFNA22 | MYO6 | 言語習得後 | 高周波・進行性 |
| DFNA25 | SLC17A8 | 言語習得後・10-50歳代 | 高周波・進行性 |
| DFNA28 | GRHL2 | 言語習得後 | 均一・緩徐に右下がり |
| DFNA36 | TMC1 | 言語習得後 | 均一・緩徐に右下がり |
| DFNA39 | DSPP | 言語習得後 | 高周波・進行性 |
| DFN41 | P2RX2 | 言語習得後 | 均一・進行性 |
| DFNA44 | CCDC50 | 言語習得後 | 低-中周波・進行性 |
| DFNA48 | MYO1A | 言語習得後 | 進行性 |
| DFNA50 | MIR96 | 言語習得後・10歳代 | 均一・進行性 |
| DFNA51 | TJP2&FAM189A2 | 言語習得後・30歳代 | 高周波・進行性 |
常染色体劣性の非症候性難聴
世界の多くの民族で,常染色体劣性の非症候性難聴者の50%にGJB2遺伝子変異がみられる[Zelante et al 1997, Estivill et al 1998, Kelley et al 1998](DFNB1を参照のこと). その他の50%の症例は他の様々な遺伝子変異が原因であり,そのうちの多くの症例は1,2家系にのみみられる難聴であった[Hilgert et al 2008] (評価手順を参照のこと).
常染色体劣性の非症候性難聴に関係のある遺伝子についての臨床症状と分子遺伝学を表4にまとめた.注:非症候性難聴の多遺伝子検査は検査手順および遺伝学的解析で述べる.
表4. 常染色体劣性の非症候性難聴の原因となる既知の遺伝子に関する臨床症状と分子遺伝学
| 遺伝子座 | 遺伝子 | 発症時期 | 聴力プロファイル |
|---|---|---|---|
| DFNB1 | GJB2 | 言語習得前1 | 通常は安定性 |
| GJB6 | |||
| DFNB2 | MYO7A | 言語習得前・言語習得後 | 非特定 |
| DFNB3 | MYO15 | 言語習得前 | 重度~最重度・安定性 |
| DFNB4 | SLC26A4 | 言語習得前・言語習得後 | 安定性・進行性 |
| DFNB6 | TMIE | 言語習得前 | 重度~最重度・安定性 |
| DFNB7/11 | TMC1 | ||
| DFNB8/10 | TMPRSS3 | 言語習得後2・言語習得前 | 進行性・安定性 |
| DFNB9 | OTOF | 言語習得前 | 通常は重度~最重度・安定性 |
| DFNB12 | CDH23 | 言語習得前 | 重度~最重度・安定性 |
| DFNB16 | STRC | 言語習得前 | 重度~最重度・安定性 |
| DFNB18 | USH1C | 言語習得前 | 重度~最重度・安定性 |
| DFNB21 | TECTA | 言語習得前 | 重度~最重度・安定性 |
| DFNB22 | OTOA | 言語習得前 | 重度~最重度・安定性 |
| DFNB23 | PCDH15 | 言語習得前 | 重度~最重度・安定性 |
| DFNB24 | RDX | 言語習得前 | 重度~最重度・安定性 |
| DFNB25 | GRXCR1 | 言語習得前 | 重度~最重度・安定性 |
| DFNB28 | TRIOBP | 言語習得前 | 重度~最重度・安定性 |
| DFNB29 | CLDN14 | 言語習得前 | 重度~最重度・安定性 |
| DFNB30 | MYO3A | 言語習得前 | 重度~最重度・安定性 |
| DFNB31 | DFN31 | 言語習得前 | ― |
| DFNB32/82 | GPSM2 | 言語習得前 | 重度~最重度・安定性 |
| DFNB35 | ESRRB | 不明 | 重度~最重度 |
| DFNB36 | ESPN | 言語習得前 | ― |
| DFNB37 | MYO6 | 言語習得前 | ― |
| DFNB39 | HGF | 言語習得前 | 重度~最重度・右下がり |
| DFNB49 | MARVELD2 | 言語習得前 | 中等度~最重度・安定性 |
| DFNB53 | COL11A2 | 言語習得前 | 重度~最重度・安定性 |
| DFNB59 | PJVK | 言語習得前 | 重度~最重度・安定性 |
| DFNB61 | SLC26A5 | 言語習得前 | 重度~最重度・安定性 |
| DFNB63 | LRTOMT | 言語習得前 | 重度~最重度・安定性 |
| DFNB67 | LHFPL5 | 言語習得前 | 重度~最重度・安定性 |
| DFNB73 | BSND | 言語習得前 | 重度~最重度・安定性 |
| DFNB76 | SYNE4 | 言語習得前 | 高周波・進行性 |
| DFNB77 | LOXHD1 | 言語習得後 | 中度~最重度・進行性 |
| DFNB79 | TPRN | 言語習得前 | 重度~最重度・安定性 |
| DFNB84 | PTPRQ | 言語習得前 | 中度~最重度・進行性 |
- 言語習得前難聴には先天性難聴も含む.
- DFNB8難聴の発症は言語習得後(10~12歳)であるが,DFNB10難聴の発症は言語習得前(先天性)である. この様な表現型の違いが遺伝型の違いに反映している.DFNB8の原因となる変異はスプライス部位の変異であり,非効率なスプライシングにより,正常な蛋白質の量を減少させていることが考えられる.この蛋白質の量は言語習得前難聴の予防には十分であるが,最終的な難聴の発症を防ぐためには十分ではない.
X連鎖性の非症候性難聴
DFNX3の特徴は伝音難聴と感音難聴の混合型であり,伝音難聴はあぶみ骨固定が原因である.他の伝音難聴とは対照的に外科的矯正は適応外である.なぜなら卵円窓に穴があいたり除去されたりすると,脳脊髄液と外リンパとの間の異常な連結が起こり, 漏出(外リンパ瘻)と完全な難聴を引き起こすからである.
その他のX連鎖性の非症候性難聴の表現型には,最重度の言語習得前難聴DFNX2や中度から最重度の幅広い言語習得後難聴のDFN4[Weegerink et al 2011],5~7歳から始まり成人期に至るまでに全周波数での重度~最重度の難聴に進行する両側性高周波難聴DFNX6が含まれる.DFN5,DFN7,DFN8の遺伝子座に関しては,まだ結果が報告されていない.
X連鎖性の非症候性難聴の原因となる遺伝子に関する臨床症状と分子遺伝学を表5にまとめる.
注:非症候性難聴の多遺伝子検査は検査手順および遺伝学的解析で述べる.
表5. X連鎖性の非症候性難聴の臨床症状と分子遺伝学
| 遺伝子座 | 遺伝子 | 発症 | タイプおよび重症度 | 周波数 |
|---|---|---|---|---|
| DFNX1 (DFN2) | PRPS1 | 言語習得後 | 感音難聴は進行性・重度~最重度 | 全周波数 |
| DFNX2(DFN3) | POU3F4 | 言語習得前 | 進行性・混合性・多様だが重度~最重度 | 全周波数 |
| DFNX4(DFN6) | SMPX | 言語習得後 | 感音難聴は進行性・中度~最重度 | 全周波数 |
非症候性難聴および難聴とミトコンドリア性難聴
ミトコンドリア遺伝子における変異の大多数により,幅広い範囲における母親由来の遺伝性多系統疾患が起こる.しかし,主にMT-RNR1 遺伝子やMT-TS1遺伝子といった遺伝子変異により非症候性難聴が起こるが,その発症機序はまだ分かっていない[Fischel-Ghodsian 1998] (表6およびミトコンドリア性非症候性難聴を参照のこと).
MT-RNR1 遺伝子は12SリボゾームRNAをコードする.この遺伝子における変異のひとつである1555G>Aは,母親由来の遺伝性非症候性難聴の原因となることが多い. 1555G>A 変異がみられる患者の中には,適切な量のアミノグリコシドの投与により難聴が誘発される場合がある.しかし,表現型は非常に多様であり,これは調節遺伝子の効果が様々であるためである.[Kokotas et al 2007].
MT-TS1遺伝子はトランスファーRNASer(UCN)をコードする. この遺伝子の7445番目の塩基におけるAからGへの置換について,ヘテロプラスミーがみられる2家系が確認されている.しかし,難聴の浸透率は低く,MT-TS1遺伝子変異自体は難聴の発症原因としては大きな役割を果たしていないことが示された.
MT-CO1はシトクロムC酸化酵素サブユニット1をコードする. 重度から最重度の難聴6人でMT-CO1の7444番目のGからAへの塩基置換のホモプラスミーとMT-RNR1の1555A>G変異が同定された[Pandya et al 1999]. 6人中5人は母系遺伝で2人はアミノグリコシド抗生剤使用歴を認めた. MT-RNR1 1555A>Gに伴う様々な難聴とは対照的に,この重複変異を持つ患者は全て重度-最重度の難聴で、完全浸透を示した.
表6. ミトコンドリア性の非症候性難聴
| 遺伝子 | 変異 | 重症度 | 浸透度 |
|---|---|---|---|
| MT-RNR1 | 961の様々な変異 | 様々 | 極めて多様・アミノグリコシド誘発性 |
| 1494C>T | |||
| 1555A>G | |||
| MT-TS1 | 7445A>G | 極めて多様 | |
| 7472insC | |||
| 7510T>C | |||
| 7511T | |||
| MT-CO1 | 7444G>A | 重度~最重度 | 完全・アミノグリコシド及びMT-RNR11555A>G変異と関連 |
検査手順
個々の患者の難聴の原因の正しい診断は,予後に関する情報を与えるものであり,正確な遺伝カウンセリングのために不可欠なものである.通常,以下の手順で行う.家族歴. 3世代にわたる家族歴を,家系内の他の難聴者に注意しつつ聴取し,関連所見を得ること.家系内近親の有益な所見の記録は,その近親本人への検査や,聴力図,耳鼻科学的評価,分子遺伝学的検査といった医療カルテの閲覧により得られる.
臨床検査. 原因不明の難聴者に対しては,必ず症候性難聴の関連所見を評価すること.見逃してはならない所見は,鰓裂の穴,嚢胞,瘻,耳介前部の孔,眼角隔離症,虹彩異色症,前頭部白髪,色素異常,高度の近視,色素性網膜症,甲状腺腫,頭蓋顔面異常である.常染色体優性の症候性難聴の発現度にはばらつきが生じやすいため,正確な診断は発端者や他の家系近親への注意深い身体的検査に基づく.
聴覚学的所見. 聴力の状態はどの年齢でも測定できる(定義を参照のこと).
- 進行性難聴者に対しては,Alport症候群, Pendred症候群, Stickler症候群への評価と,側頭骨のCT画像診断を行うこと.
- 突発性難聴や進行の速い難聴は,側頭骨異常(Pendred症候群および鰓弓耳腎症候群),腫瘍(神経線維腫症2型),免疫関連難聴や,外傷,感染(梅毒,ライム病),代謝異常,神経学的異常,循環障害を伴うことがある.
- WFS1遺伝子変異は,高周波の聴力は維持されて主に低周波が損なわれる優性遺伝性の難聴の家系の75%にみられる[Cryns et al 2003].
側頭骨のCT画像診断. 側頭骨のCT画像診断は内耳の先天性異常(Mondini型異形成,Michel型無形成,前庭水管肥大症・拡大症,内耳道拡大症)を検出する際に役立つため,進行性難聴者に対しては考慮すること.
内耳障害は以下の変異に関連があるため,CT画像診断による側頭骨異常の検出により,どのような分子遺伝学的検査 を行うべきかの手掛かりとなる.
- SLC26A4 変異(Pendred症候群を参照のこと).
- POU3F4 変異[Vore et al 2005]
検査
感音難聴の乳児に対しては,サイトメガロウイルス(CMV)検査を考慮する必要がある.子宮内におけるCMV曝露は,新生児期のCMV抗体価の上昇が検出されるか尿培養で陽性反応が検出されれば診断される.これらの検査はもっと後でも可能だが,出生後に獲得されたCMV感染の可能性が出てくるために結果の解釈が混同される.出生後のCMV感染は珍しくないが,難聴とは無関係である.
分子遺伝学的検査.
極端な遺伝的異質性や表現型のばらつきは, 1遺伝子ずつのスクリーニング法を用いた非症候性難聴の遺伝学的診断を難しくする。このため、いくつかのグループにより多遺伝子スクリーニングパネルが開発された. これらのスクリーニングパネルは研究室によって用いる手法や解析する遺伝子数が変動していることに注意を要する.いくつかの研究室では数遺伝子の既報告変異のみを標的とし, 一方で他の研究室では非症候群性難聴に関連するすべての遺伝子をシークエンスする. このような検査がより浸透していけば, 遺伝性難聴の管理が全てのタイプの難聴に対する包括的遺伝子検査へと変化していくだろう.
1遺伝子検査が実施される際は、疫学や表現型データに基づいて優先して検査する遺伝子が決まる。例えば、GJB2 遺伝子(コネキシン26蛋白をコードする遺伝子)とGJB6遺伝子(コネキシン30蛋白をコードする遺伝子:DFNB1, 分子遺伝学的検査を参照)に対する分子遺伝学的検査は常染色体劣性遺伝形式またはDFNB1の「擬似優性遺伝」形式に一致した先天性の非症候性感音難聴者の評価の際に考慮する.擬似優性遺伝とはある家系で2世代以上に発現する常染色体劣性疾患を指す.このような遺伝形式は一般集団における保因者率が高い場合に起こりやすい.GJB2遺伝子やGJB6遺伝子に対する分子遺伝学的検査は,2世代にわたり発症がみられる非症候性難聴家系に実施すべきである.
常染色体劣性の非症候性難聴であると考えられる先天性の高度―最重度の難聴児にDFNB1遺伝子座のGJB2遺伝子やGJB6遺伝子の変異がみられない場合,お座りや歩行といった運動発達遅滞がみられる場合は,Usher症候群1型を考慮する.
側頭骨のCT検査で前庭水管肥大症・拡大症やMondini型異形成を認めた時はSLC26A4の変異スクリーニングを実施すべきである(Pendred症候群を参照).
常染色体優性非症候群性難聴の家系では、聴力プロファイルから可能性のある遺伝子を選択した後に行われる変異に対するスクリーニング検査は有益である(表3を参照).このアプローチを容易にするためのコンピューターアルゴリズムが開発中である[Hildebrand et al,2008].
これらの新しいアプローチでも診断が得られない場合は、多遺伝子パネルを用いた非症候群性難聴の全遺伝子スクリーニングで結果が得られるからもしれない[Shearer et al 2010]
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
遺伝性難聴は常染色体優性,常染色体劣性,X連鎖性の遺伝形式をとりうる.難聴を伴うミトコンドリア異常症も発症する.
近親者のリスク― 常染色体優性遺伝性難聴
発端者の両親
- 常染色体優性の難聴と診断された者の大多数は親の一人が難聴である.家族歴が陰性であることはほとんどない.
- 常染色体優性の難聴の発端者は新生突然変異の結果として遺伝性難聴を発病した可能性がある.新生突然変異による症例の割合は不明であるが低いと考えられる.
- 一見して新生突然変異であるような発端者の両親に対して勧められる検査には聴力検査と分子遺伝学的検査がある.
注: 常染色体優性の遺伝性難聴と診断された者には親の1人が難聴である場合ほとんどであるが,家系内の難聴に気づかなかった場合,親の発症が遅い場合,無症状の片親における変異アレルの浸透率が低下した場合,遺伝性難聴の新生突然変異が生じた場合には,家族歴が陰性となることがある.
発端者の同胞
- 同胞のリスクは発端者の両親の遺伝状態による.
- 発端者の両親の一人が変異アレルを持つ場合,同胞が変異アレルを受け継ぐリスクは50%である.
- それぞれの症候群により,臨床上の重症度や疾患の表現型は同一の変異を持つ患者の間でも異なることがある.従って,発症年齢や疾患の進行は予測できないことがある.
発端者の子
- 常染色体優性の遺伝性難聴者では,子一人あたりに変異アレルを伝える確率は50%である.
- それぞれの症候群により,臨床上の重症度や疾患の表現型は同一の変異を持つ患者の間でも異なることがある.従って,発症年齢や疾患の進行は予測できないことがある.
一見,新生突然変異とみられる家系での配慮.
常染色体優性疾患の発端者の両親のどちらにも難聴の原因となる変異や疾患の臨床症状がみられない場合,発端者が新生突然変異を持つ可能性がある.しかし,生物学的父親や母親が異なる場合(生殖補助医療など)や非公開の養子縁組などの疾患とは無関係な理由も考えられることもあるだろう.
近親のリスク―常染色体劣性遺伝性難聴
発端者の両親
- 両親は絶対的ヘテロ接合体であるため,難聴の原因となる変異を1コピー持つ.
- ヘテロ接合体は無症状である.
発端者の同胞
- 受精時に,子が難聴となる確率は25%であり,正常な聴覚を持つ保因者となる確率は50%,正常な聴覚を持ち保因者ともならない確率は25%である.
- リスクのある同胞が正常な聴覚を持つと分かった場合,この同胞が保因者である確率は2/3である.
- ヘテロ接合体は無症状である.
- それぞれの症候群により,臨床上の重症度や疾患の表現型は同一の変異を持つ患者の間でも異なることがある.従って,発症年齢や疾患の進行は予測できないことがある.
- GJB2遺伝子関連の重度~最重度の難聴の発端者については,GJB2遺伝子について同一の遺伝子型を持つ同胞が,重度~最重度の難聴となる確率は91%,軽度~中等度の難聴となる確率は9%である.
- GJB2遺伝子関連の軽度~中等度の難聴の発端者については,GJB2遺伝子について同一の遺伝子型を持つ同胞が,軽度~中等度の難聴となる確率は66%,重度~最重度の難聴となる確率は34%である.
発端者の子
すべての子は絶対的保因者となる.
発端者の他の近親
絶対的ヘテロ接合体の同胞がヘテロ接合体である確率は50%である.
家系メンバーのリスク― X連鎖性遺伝性難聴
発端者の両親
- X連鎖性難聴男性の父親は難聴にはならず,変異の保因者ではない.
- X連鎖性難聴の息子を一人と,その他にX連鎖性難聴の男性近親を持つ女性は,絶対的ヘテロ接合体である.
- 家系分析で難聴男性が家系内で唯一の難聴者である場合,難聴者の母親の保因者状態に関して幾つかの可能性を考慮する必要がある.
- 難聴男性が新生突然変異で難聴を引き起こす変異を持ち,母親は保因者でない場合;
- 難聴男性の母親が新生突然変異で難聴を引き起こす変異を持つ場合,(a) 「生殖細胞変異」 (すなわち,母親自身の受精時に起こった変異であり,母親の身体の全細胞に存在する変異),もしくは(b)「生殖細胞モザイク」(すなわち,母親の生殖細胞の幾つかにのみ存在する変異)のどちらかが考えられる.
- 難聴男性の祖母が新生突然変異により難聴を引き起こす変異を持った場合.
- しかし,新生突然変異の頻度や母親の生殖細胞モザイクの起こる確率や頻度に関するデータはない.
発端者の同胞
- 同胞のリスクは発端者の母親の遺伝状況による.
- 保因者である女性が難聴を引き起こす変異を各妊娠で伝える確率は50%である.
- 変異を受け継いだ息子は難聴となる.変異を受け継いだ娘は保因者となり,聴覚は正常である可能性が高い.
- 母親が保因者でない場合,同胞のリスクは低いが,生殖細胞モザイクの可能性があるために一般集団よりは高い.
- それぞれの症候群により,臨床上の重症度や疾患の表現型は同一の変異を持つ患者の間でも異なることがある.従って,発症年齢や疾患の進行は予測できないことがある.
発端者の子
X連鎖性の遺伝性難聴男性は,難聴の原因となる変異を全ての娘に伝えるが,息子には伝えない.
発端者の他の近親
発端者の母方のおばは保因者であるリスクがあり,おばの子孫はその性別により保因者となるリスクや難聴となるリスクを持つ場合がある.
近親のリスク― 難聴を伴うミトコンドリア異常症の特徴
発端者の両親
- 発端者の母親は(通常)ミトコンドリア変異を持ち,症状を呈する場合と呈していない場合がある.
- 発端者の父親は発病性のミトコンドリアDNA変異を持っているリスクはない.
- あるいは,発端者が新生突然ミトコンドリア変異を持つ可能性がある.
発端者の同胞
- 同胞のリスクは母親の遺伝状況による.
- 母親がミトコンドリア変異を持つ場合, 全ての同胞に変異を受け継ぐリスクがある.
発端者の子
- ミトコンドリアDNA変異を持つ女性の全ての子は変異を受け継ぐリスクがある.
- ミトコンドリアDNA 変異を持つ男性の子はリスクがない.
発端者の他の近親.
他の家系近親のリスクは発端者の母親の遺伝状況による. 母親がミトコンドリア変異を持つ場合,母親の同胞や母親自身の母親もリスクがある.
近親のリスク―経験的リスク
特定の診断がつかない場合(や遺伝形式が確定しない場合),次のような経験的所見が用いられる.
夫と妻が難聴で難聴の子どもが一人いて,難聴の家族歴が陰性の場合,次の子どもが難聴となる経験的確率は18% である[Green et al 1999].
- GJB2遺伝子およびGJB6遺伝子の分子遺伝学的検査により難聴児がDFNB1型ではない場合には,コネキシン26に関係のない難聴の再発リスクは14%である[Green et al 1999].
- 難聴の夫婦が同族結婚もしくは血族結婚の割合が高い集団の出身者である場合には,常染色体劣性の遺伝形式の可能性が高まるために,次の子どもが難聴となる確率は25%近くになる.
難聴者と難聴者でない者の間に生まれた子どもが難聴となる経験的確率は10%である [Green et al 1999].
- リスクのほとんどは常染色体優性の症候性難聴に起因する.
- 症候性難聴と常染色体劣性の遺伝形式の家族歴が除外された場合,難聴リスクは主に擬似優性の劣性難聴の発症率に関わってくる.GJB2遺伝子検査によりこのリスクの多くが確認できる.
常染色体優性の難聴が除外されている非血族結婚の難聴夫婦の子が難聴となる経験的リスクは約15%である [Green et al 1999].
- 両親ともGJB2遺伝子関連難聴である場合,子のリスクは100%である.
- 両親が異なる2つの遺伝子座の変異による常染色体劣性の難聴だとわかっている場合,子が難聴となる確率は一般集団よりも低くなる.
(常染色体劣性の非症候性難聴であると考えられる)発端者の難聴者の正常な聴覚を持つ同胞と,難聴者との間の子が難聴となる経験的リスクは1/200 (0.5%)であり,これは一般集団のリスクの5倍である
GJB2遺伝子およびGJB6遺伝子に対する分子遺伝学的検査により,リスクが高まるかどうか明らかにすることが可能である.聴覚が正常な同胞がGJB2遺伝子変異やGJB6遺伝子変異の保因者であり,同胞のパートナーがDFNB1型の難聴者である場合には,難聴児が生まれる確率は50%である.
遺伝カウンセリングに関連した問題
難聴者とのコミュニケーションには熟練した通訳が必要である.
難聴者は難聴を個性でありハンディキャップや障害,「治療」や「治癒」を要する病気,もしくは「予防」すべき病気であると考えていないこともある.
難聴の予防や出産,家族計画についての情報ではなく,医学的,教育的,社会的サービスに関する情報を含めた難聴の原因について情報を得たがっている難聴者が多い.すべての遺伝カウンセリングの場合と同様であるが,カウンセラーが難聴者および家族の質問,心配,恐れを確認,理解し,尊重することが重要である[Middleton et al 1998, Arnos 2003].
用語の選択に関して,注意すること.「リスク」よりも「確率」を,「聴覚障害」よりも「難聴」を用いる方がよい.「罹患」,「異常」,「発病性」といった用語は用いないこと.
家族計画
- 遺伝的リスク,保因者状態,出生前診断の利用の有無を確認する最適な時期は妊娠前である.
- 若い成人難聴者に遺伝カウンセリング(子孫へのリスクや出産手段に関する話し合いを含む)を勧めることは適切である.
DNAバンキングは,将来の使用のために,(通常は白血球から調整した)DNAを貯蔵しておくことである.検査手法や,遺伝子,変異,疾患への理解は将来改善する可能性があり,難聴者のDNAを貯蔵しておくことは考慮されるべきである.ことに,現在行っている分子遺伝学的検査が研究段階でのみ実施されている場合には有意義である.このサービスを行っている機関については DNA bankingの項を参照のこと.
出生前診断
幾つかのタイプの遺伝性難聴に対する出生前診断は,通常胎生週数15~18週頃に実施される羊水検査や胎生週数10~12週頃に実施される絨毛生検(CVS)により採取された胎児細胞のDNA分析により可能である.出生前診断の実施前に,家系内の難聴者に難聴の原因となるアレルが同定されていなければならない.
注:胎生週期とは最終月経の第1日から換算するか,超音波による計測によって算出される.
難聴のような疾患に対する出生前診断の要望はあまり多くない.出生前診断を行うことに対しては,専門医の間でも家族によっても考え方が異なるだろう.特に,検査が妊娠中絶を考慮したうえで行われる場合には尚のことである.大抵の医療機関では出生前診断を受けるかどうかの決定は両親の選択に委ねると考えるであろうが,この問題について議論を行うことが望ましい.
着床前診断 (PGD) 着床前診断は家系内に難聴の原因となる変異が同定されている場合に実施可能である.着床前診断を行っている施設に関しては [GENETEST]を参照のこと.
臨床的マネジメント
症状の治療
理想的には,難聴者を評価し治療するチームは,小児初期の耳鼻科疾患の管理に熟達した耳鼻咽喉科医,小児の難聴評価の経験が豊富な聴覚士,臨床遺伝医,小児科医で構成されるべきである.難聴者教育の専門家,神経科医,小児眼科医も必要とされる場合もある. 評価の中で重要な点は,適切なハビリテーション機器の選択である.聴覚補助器具,振動補聴器,人工内耳が考えられる.人工内耳は生後12カ月以降の重度~最重度の難聴児に対して考慮される.
DFNB1遺伝子座におけるGJB2遺伝子およびGJB6遺伝子に変異がみられる重度~最重度の常染色体劣性の先天性非症候性難聴の子どもに対して人工内耳が選択された場合,その効果は傑出している[Bauer et al 2003].
一次病変の予防
進行性感音難聴と進行性運動失調がみられる子どもに対しては,神経学的徴候や皮膚症状の有無にかかわらず,必ずビオチニダーゼ欠損症を考慮し,できるだけ早く不可逆的な後遺症が起こる前に治療を開始すること.
続発性合併症の予防
どのような発症原因であれ,難聴を放置しておくと続発症が発生する.2歳以前の聴覚障害は低い読解力,低いコミュニケーション能力,低い発話能力との関連性が認められている.
教育的介入だけではこれらの障害を十分に修正するためには足りない.対照的に,補聴器,耳鼻科手術,人工内耳といった聴覚的介入を早期に行うことには有効性が認められている [Smith et al 2005].
認知能力,数学能力,読解能力の低下は難聴と関連性があるが,遺伝性難聴者への調査から,これらの能力不足は難聴原因とは本質的に結びついていないことが分かった.例えば,GJB2遺伝子関連の難聴者の認知能力を測定したところ,人工内耳埋込術後ではヒスキーIQ,読解能力とも正常であった[Bauer et al 2003]. 従って,難聴の早期発見と適切な時期の介入が,言語習得前難聴の小児の最適な認知発達にとっては非常に重要である.
経過観察
継続的な聴覚検査の実施が不可欠である.
- 難聴の不変・進行を記録.
- 中耳浸出といったその他の難聴原因の確認および治療.
SLC26A4遺伝子変異による常染色体劣性の非症候性難聴者では,難聴は進行性であるため,1年に1回聴覚検査を行う必要がある.また,ペンドレッド症候群に合致する診断がつけられた場合には,甲状腺機能も追跡すること.
回避すべき薬剤・環境
騒音への曝露は難聴の原因となるよく知られた環境的要因の一つである.このリスクは回避により最小限とすることができるために,難聴であることが分かっている者は適切にアドバイスを受けること.
リスクがある近親の検査
最低限,遺伝性難聴のリスクを持つ全ての子どもはスクリーニングのための聴力検査を受ける.
遺伝カウンセリング目的で行われるリスクのある近親に対する検査に関連する諸問題については,遺伝カウンセリングを参照のこと.
研究段階の治療
現在行われている難聴者へのハビテーションは,聴覚補助具や人工内耳埋込術による増幅が中心である.研究段階の治療には,発話を電子的でかつ音響学的に処理する聴覚補助具と接続した短い人工内耳電極の利用,両耳埋め込み術がある[Gantz et al 2005].
種々の疾患に対する臨床試験についてはClinicalTrials.govを参照のこと.
更新履歴
- Gene Review著者: Richard JH Smith, MD, Guy Van Camp, PhD
日本語訳者: 窪田美穂(ボランティア翻訳者),櫻井晃洋(信州大学医学部附属病院遺伝子診療部)
Gene Review 最終更新日: 2008.12.2. 日本語訳最終更新日: 2009.7.20 -
Gene Review著者: Richard JH Smith, MD, Guy Van Camp, PhD
日本語訳者: 窪田美穂(ボランティア翻訳者),岸本 洋子(島田療育センターはちおうじ)Gene Review 最終更新日: 2014.1.9. 日本語訳最終更新日: 2015.3.25 (in present)