コフィン・シリス症候群
(Coffin-Siris Syndrome)
[Synonyms:Fifth Digit Syndrome]
Gene Reviews著者: Samantha Schrier Vergano, MD, Gijs Santen, MD, PhD, Dagmar Wieczorek, MD, Bernd Wollnik, MD, Naomichi Matsumoto, MD, PhD, and Matthew A Deardorff, MD, PhD.
日本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日:2021.8.12. 日本語訳最終更新日: 2023.12.15.
要約
疾患の特徴
Coffin-Siris症候群(CSS)は、古典的なものでは、第5指ならびにその他の指趾の末節骨もしくは爪の無形成、種々の程度の発達ないし認知機能の遅延、特徴的顔貌、筋緊張低下、男性型多毛症/多毛症、疎な頭髪を特徴とする。発生する可能性のある先天奇形としては、心奇形、消化器系奇形、尿路性器奇形、中枢神経系奇形がある。他の所見で多くみられるものとしては、摂食障害、発育遅延、眼科的異常、聴覚障害がある
診断・検査
分子的基礎が判明する以前、CSSの診断は専ら臨床所見をもとに行われていた(合意済の臨床的診断基準が確立していたわけではない)。発端者におけるCSSの診断は、これを示唆する所見がみられることに加え、表1に示した遺伝子のうちの1つにヘテロ接合性の病的バリアントを同定することをもって確定する。
臨床的マネジメント
症状に対する治療:
最良の発達を誘導するため、作業療法、理学療法、言語治療を行う。摂食療法、栄養補給に加え、栄養管理上必要であれば胃瘻造設・チューブ留置を行う。眼科的異常や難聴に対しては通常の管理を行う。
定期的追跡評価 :
発達の進行状況、ならびに治療的・教育的介入に関する評価のため、年に1度、発達小児科医による評価を行う。摂食や体重増加の状況を把握するため、必要に応じ、消化器内科医や摂食の専門家の手で追跡評価を行う。眼科的ないし聴覚的異常に対しては、通常の追跡管理を行う。
遺伝カウンセリング
CSSは常染色体顕性遺伝の遺伝形式をとる。ただ、罹患者の大多数は、CSSを引き起こす病的バリアントがde novoで生じた例である。家系内に存在するCSSの原因となった病的バリアントがすでに同定されている場合は、高リスクの妊娠に向けた出生前検査や着床前遺伝学的検査が可能である。
診断
Coffin-Siris症候群(CSS)に関しては、今のところ公式な臨床診断基準は確立されていないものの、臨床診断を下す上で鍵となるポイントはいくつか存在する。
本疾患を示唆する所見
次のような所見を伴う例については、Coffin-Siris症候群(CSS)を疑う必要がある[Fleckら2001,Schrierら2012,Koshoら2014b,Santenら2014]。
- 第5指趾の爪ないし末節骨の低形成ないし無形成。CSSの臨床診断がなされた罹患者は、通常第5指末節骨の無形成か低形成、ないし爪の欠損を示す。爪の欠損は通常第5指であるが、他の指にもみられることがある(図1C,D,E,F)。足趾にも異常がみられることがあり、その場合、異常が複数の足趾に及ぶ傾向がみられる。
- 種々の程度の発達遅滞あるいは認知機能の遅延
- 顔面症候[Schrierら2012]。典型所見を有する罹患者では、厚く反転した上下口唇を伴う広い口、広い鼻梁と鼻尖、濃い眉、長い睫毛がみられる。これらが合わさって、CSS罹患者は粗野さを感じさせる顔となる(図1A,B)。
- 中枢神経を起源とする筋緊張低下
- 男性型多毛症ないし多毛症。あまり毛がないはずの場所(背中など)における発毛、あるいは腕や顔の多毛。
- とりわけ側頭部に顕著にみられる疎な頭髪(特に乳児期)
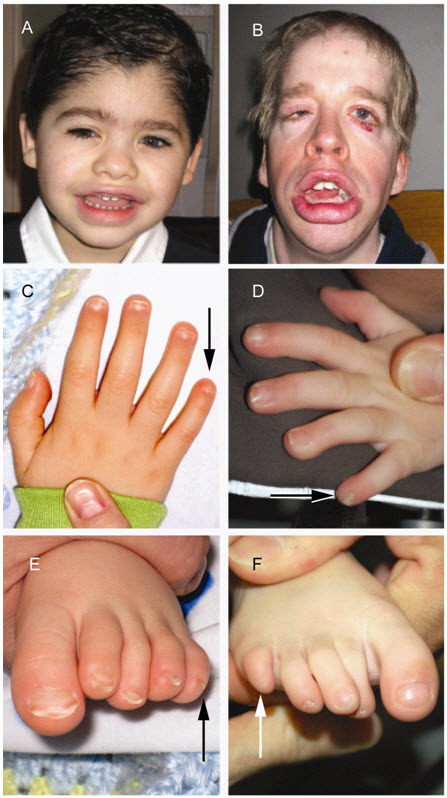
図1:Coffin-Siris症候群の古典的所見
Coffin-Siris症候群の臨床診断が下された5歳男児(A)、同29歳男性(B)にみられた顔面症候(密な眉毛、粗野な顔、厚く反転した口唇)、第5指の爪(C)、末節骨(D)の低形成、第5趾の爪(E)、趾節骨(F)の無形成
診断の確定
発端者におけるCSSの診断は、これを示唆する所見を有することに加え、表1に挙げた遺伝子の1つにヘテロ接合性病的バリアントが同定されることをもって確定する。
注:表1に挙げた遺伝子の1つにヘテロ接合性の意義不明バリアントが検出された場合、それは本疾患の診断を確定するものでも否定するものでもない。
分子レベルの検査手法としては、ふつう、マルチ遺伝子パネル検査、もしくはより網羅的なゲノム検査が用いられる。
注:もう一つの選択肢として、病的バリアントの出現頻度順に直列型で行う単一遺伝子検査も考えられる。
ただ、この表現型に関与する遺伝子がかなりの数に及ぶだけに、こうしたやり方はあまり一般的ではない。それでも、罹患者の数としては、ARID1Bの変異が表1に示した他の遺伝子の変異と比較してかなり多いので、最初にARID1Bの配列解析、次いでARID1Bの遺伝子標的型欠失/重複解析というやり方も検討に値する。もしこれで診断がつかないようであれば、次にマルチ遺伝子パネル検査もしくは網羅的ゲノム検査を行うといった順番になろう。
- マルチ遺伝子パネル検査
表1に挙げた遺伝子、ならびにその他の関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネル検査が検討対象になりえよう。
注:(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。したがって、臨床医には、どのマルチ遺伝子パネルを用いれば現状を引き起こしている原因遺伝子を同定できる可能性が高いかという点を判断することが求められる。
(3)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝学的検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
- 網羅的ゲノム検査
網羅的ゲノム検査の場合、臨床医の側で疑わしい遺伝子の目星をつけておく必要はない。エクソームシーケンシングが最も広く用いられているが、ゲノムシーケンシングを使用することも可能である。
網羅的ゲノム検査の基礎的情報についてはここをクリック。
ゲノム検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
表1:Coffin-Siris症候群で用いられる分子遺伝学的検査
| 遺伝子1 | その遺伝子の病的バリアントがCSS全体の中で占める割合2 | その手法で病的バリアントが検出される割合3 | |
| 配列解析4 | 遺伝子標的型欠失/重複解析5 | ||
| ARID1A | 5%未満 | 100%6 | 不明7 |
| ARID1B | 37%以内 | 95%以内 | 5%以内8 |
| ARID2 | 稀9 | 100% | 不明7 |
| DPF2 | 稀10 | 100% | 不明7 |
| PHF6 11 | 稀12 | 100% | 不明7 |
| SMARCA2 13 | 2%以内 | 90%超 | 1人の罹患者 |
| SMARCA4 | 7%以内 | 100% | 不明7,14 |
| SMARCB1 | 7%以内 | 100% | 不明7,14 |
| SMARCC2 | 稀15 | 75%以内 | 4人の罹患者 |
| SMARCE1 | 2%以内 | 100% | 不明7,14 |
| SOX4 | 稀16 | 100% | 不明7 |
| SOX11 | 2%以内17 | 40%以内18 | 現在までにCSSの表現型を示す欠失の罹患者が7人報告されている19。 |
| 不明20 | 40%以内 | 対象外 | |
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- 割合を示す数字は、脚注に特記したものを除き、CSSの臨床診断がなされたコホートの報告を総合して算出したものである[Tsurusakiら2012, Santenら2013, Wieczorekら2013, Tsurusakiら2014b]。
- これらの遺伝子で検出されているアレルバリアントの情報については、「分子遺伝学」の項を参照。
- 配列解析を行うことで、benign、likely benign、意義不明、likely pathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、遺伝子内の小欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。
列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。 - 遺伝子標的型欠失/重複解析では、遺伝子内の欠失や重複が検出される。具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失/重複の検出を目的に設計された遺伝子標的型マイクロアレイなどがある。
- ARID1Aに関しては、病的バリアントのモザイク例の報告がある[Santenら2013,Wieczorekら2013]。
- 遺伝子標的型欠失/重複解析の検出率に関するデータは得られていない。
- これまでに、ARID1Bを含む6q25.3領域の小欠失が、以下の例で報告されている。
(a)CSSの分子的基盤が解明される前の段階で確認がなされた複数の子ども[Tsurusakiら2012]
(b)ARID1Bを含む小欠失が先に検出された後になって、CSS類似の所見が認められた複数の子ども[Santenら2012]
(c)臨床症候だけではCSS類似例とまでは断定できないものの、わずかに、あるいはさまざまな幅で症候群的要素を併せもつ知的障害の罹患者[Nagamaniら2009,Halgrenら2012,Hoyerら2012,Michelsonら2012]
こうした欠失の罹患者は、ARID1Bの周囲の遺伝子がどの範囲まで欠失しているかによって、さまざまな複合的な症候を示すことがあることに注意が必要である。 - この遺伝子の病的バリアントをもつ罹患者で、これまでに報告されているのは、7人のみである[Gazdaghら2019]。
- この遺伝子の変異が検出されているのは、これまでに10人未満である[Miloneら2020]。
- 幼少時にCSSの診断を受けた人の中に、その後、PHF6の病的バリアントが同定された例が複数みられる。ただ、そうした例は、年齢を重ねるに従って、むしろBorjeson-Forssman-Lehmann症候群のほうにより近い顔貌になっていっている[Wieczorekら2013]。 「鑑別診断」の項を参照されたい。
- CSSの表現型を呈する罹患者で、この遺伝子の変異が報告されているのは、これまでに2例のみである[Wieczorekら2013]。
- 当初、CSSと考えられた罹患者を再評価した結果、むしろNicolaides-Baraitser症候群のほうに一致点が多いという結論に至っている[Tsurusakiら2012,Van Houdtら2012]。ただ、SMARCA2の病的バリアントを有する罹患者の相当数が、当初はCSSの診断を受けているため、一応、著者らはこれも含めることとした。 「鑑別診断」の項を参照されたい。
- SMARCA4、SMARCB1、SMARCE1の病的バリアントは、いずれも異常機能獲得型変異であるとのデータがあり、このことから考えて、大きな病的欠失や重複が生じている例は考えにくい。それでも、関連ドメインに生じるインフレーム欠失やインフレーム重複が病的なものとなる可能性は考えられる。 SMARCA4で、その種の欠失が報告されている(「分子遺伝学」の項を参照)。
- CSSに一致すると言うこともできる症候を有する例で、この遺伝子の変異がこれまでに15例報告されているものの、古典的な症候を有するのは4例のみである[Macholら2019]。
- CSSの表現型を有する罹患者で、この遺伝子の変異が報告されているのは、これまで4例に過ぎない。
- Tsurusakiら[2014a]
- Tsurusakiら[2014a],Hempelら[2016]
- Hempelら[2016]
- CSSの罹患者で、これまでに知られているいずれの遺伝子にも病的バリアントが同定されていない例が、約40%に上る[Tsurusakiら2012,Santenら2013,Wieczorekら2013,Tsurusakiら2014b]。
核型分析ならびに染色体マイクロアレイ検査(CMA)
先天奇形や発達遅滞を有する罹患者の多くは、最初の評価手段の一部として、核型分析ないしCMAを受けるはずである。もし、CSSの症候を有しながらも、これまでに述べたような検査で病的バリアントが検出されなかった罹患者があれば、稀なゲノム再編成を念頭に、核型検査やCMAの施行を考慮すべきである。実際にCSSでゲノム再編成がみられた例が報告されている[Backxら2011,Halgrenら2012,Malliら2014]。
臨床的特徴
臨床像
以下に記載する情報は、Coffin-Siris症候群国際協力機構からの2つの報告[Koshoら2014b,Santenら2014]にあるデータをまとめたものである。このセクションでは、分子レベルで分けられる各サブタイプに共通してみられる所見に焦点を当てることとし、出現頻度にばらつきがある所見や、遺伝子上の各病因間での重症度の違いなどについては、「遺伝型-表現型相関」の項で述べる。
初期にみられる特徴
出生前
出生前は、成長も正常範囲内にあり、特段の所見を示さないのがふつうである。稀に、中枢神経系奇形や心奇形、子宮内発育遅延、小頭症がみられることもある。
新生児期/乳児期
Coffin-Siris症候群(CSS)患児の多くは、出生時においては臨床的にCSSと同定されないものの、それでもいくつかの先天奇形が確認されることがある。
- 第5指趾/爪の低形成。
大半の患児は少なくとも第5指趾の短指趾(患児の65%)、ならびに1つないしそれ以上の数の爪の低形成(80%)を示す。ただ、分子レベルでCSSと確認された罹患者の中にも、第5指趾の異常がほとんどあるいは全くみられない例があるので注意を要する。
- 顔面の形態異常(出生時は30%以内)。
顔貌は、年齢とともに粗野さを増す。したがって、もっと後の小児期まで、特徴的顔貌は表面に現れないことがある。
- 男性型多毛がしばしばみられる。
- 中枢神経系、心臓、泌尿生殖器系の奇形(「小児期の所見」を参照)。
- 乳児期にみられる上記以外の次のような所見がCSSの最初の徴候であることもある。
- 摂食障害(90%)や成長の遅れ
- 筋緊張低下(75%)
- てんかん発作(50%)
- 聴力障害(45%)
- 視覚障害(40%以内)
小児期の所見
発達遅滞
発達遅滞/認知機能の遅延が表面化するのは、発達指標に遅れが認められたとき、ないし正式な認知検査が施行されたときであるのがふつうである。
- 平均的には、CSS患児のお座りは12ヵ月、歩行開始は30ヵ月、初語は24ヵ月である。
- 受容言語に比して表出言語のほうにより重度の影響が現れ、少なからぬ割合の罹患者はスピーチの表出が全くみられない状態である。
- 知的障害が大部分にみられ、その程度は、ふつう中等度から重度(IQの範囲は40から69)であるが、IQ値が97という例も報告されている[Santenら2012]。
- 行動の異常としては、多動(10%近く)、攻撃性(10%近く)に加え、時として自閉症の症候もみられる。
脳/中枢神経系の問題
- Dandy-Walker奇形、単純脳回、脳梁無形成といった中枢神経系奇形
- てんかん発作ならびにチック。さまざまな種類のてんかん発作が報告されている。てんかん発作やチックの初発年齢について、特定の年齢はない。
- 乳児期にみられる筋緊張低下(75%)はふつう持続性である。
顔面所見(図1参照)
- 粗野な顔(95%)
- 濃い眉(90%)
- 長い睫毛(85%)
- 平坦な鼻梁(50%)
- 短い鼻(50%)
- 上向きの鼻孔(50%)
- 広い鼻尖(75%)
- 広い鼻底(50%)
- 厚い鼻翼(70%)
- 広い人中(70%)
- 広い口(80%)
- 薄い上赤唇(50%)
- 厚い下赤唇(80%)
筋骨格系所見
- 小さな第5指趾の爪(80%)
- 彎指趾(40%)
- 骨年齢の遅延(40%)
- 関節弛緩(66%)
- 脊柱側彎(30%)
- ヘルニア(10%)
皮膚・毛髪所見
- 多毛症(95%)が、罹患者の帰属する民族集団においてふつうありえない部位(例えば背中や肩)に生じる。
- 低い前頭毛髪線が多くみられる(75%)。
- 疎な頭髪(60%)
頭髪はふつう通りの時期に出現するが、頭髪自体は非常に細い。
摂食障害
明らかな腸の奇形がみられないにもかかわらず、食物嫌悪や摂食困難を呈する子どもがいる。
成長の問題
- 身長、体重は大多数の罹患者で50パーセンタイルを下回り、20%については5パーセンタイルを下回る。
- 骨年齢は、概して暦年齢に対し2,3年の遅れを示す。
- 歯牙年齢が遅延する(40%)。
聴力障害
聴力障害(45%)が、しばしば反復性の上気道感染症に伴う形でみられる。
眼科的異常
- 眼瞼下垂(50%)
- 斜視(50%)
- 近視(20%)
感染症の頻発(60%)
これについては、特にこれといった特徴はないものの、上気道のウイルス性感染症が比較的多くみられる。
奇形
- 心室中隔欠損、心房中隔欠損、Fallot四徴、動脈管開存などの心奇形(35%)
- 腎奇形、尿性器奇形(35%近く)。内訳は、停留精巣が最も多くみられるものの、馬蹄腎、尿道下裂、その他の奇形もみられる。
腫瘍リスク
CSSを引き起こす遺伝子の一部の病的バリアントについて、腫瘍発生との関連が指摘されている(「癌ならびに良性腫瘍」の項を参照)ものの、CSSの腫瘍リスクに関しては、まだデータが不足している現状である。CSS罹患者3例で腫瘍が報告されている。
- ARID1Aの病的バリアントを有する8例中1例で肝芽腫が報告されている[Tsurusakiら2012,Koshoら2014b]。
- ARID1Bを含む14の遺伝子にまたがる4.2-Mbの欠失を有する1罹患者で、甲状腺乳頭癌の発生をみている[Vengoecheaら2014]。
- CSSの1罹患者について、多発性神経鞘腫(schwannomatosis)、ならびにSMARCB1の病的バリアントを報告した複数の研究がみられる[Carterら2012,Schrierら2012,Gossaiら2015]。
予後
長期的報告が存在しないため、Coffin-Siris症候群罹患者の寿命についての情報は得られていない。誤嚥性肺炎やてんかん発作による子どもの死亡報告がみられるものの、これは多くみられるものではない[Schrierら2012]。CSS罹患者の予後をよりよく調べるためのCoffin-Siris症候群国際コンソーシアム[Koshoら,2014a]による調査が進行中である。
遺伝子ごとにみた表現型との相関
CSSの臨床診断が下された罹患者で、ARID1A、ARID1B、ARID2、DPF2、SMARCA4、SMARCB1、SMARCC2、SMARCE1、SOX4、SOX11の病的バリアントを伴う例について、遺伝子ごとにみた表現型との相関がわかっている[Wieczorekら2013,Koshoら2014b,Santenら2014,Tsurusakiら2014a,Hempleら2016,Vasileiouら2018,Gozdaghら2019,Macholら2019,Zawertonら2019]。
ARID1B
ARID1Bの病的バリアントを有する罹患者は、概してCSSのスペクトラムの中にあって最軽症の状態を示し、正常な成長を示すことが多い。
3分の2はある程度重度の部類の摂食障害を、3分の1はてんかん発作を、3分の1は脳梁低形成を示す。顔面の特徴はCSSで普通にみられる通りのものであるが、時に軽度のこともある。
ARID2
出生時の異常を伴わない罹患者が多い。
SMARCA4
SMARCA4の病的バリアントを有する罹患者は、出生前は軽度、出生後は軽度から中等度の成長障害を示し、吸啜/摂食障害がほぼ全例でみられる。重度の発達遅滞を呈する罹患者も時にみられるものの、特徴としては、むしろ行動の問題のほうが大きく現れる傾向にある。顔貌の粗野さは少なめながら、第5指趾とその爪の低形成は必発に近い(他の指趾の爪の低形成も時にみられる)。症例によっては、指趾節間関節や末節骨の隆起がみられることもある。
SMARCB1
SMARCB1の病的バリアントを有する罹患者は、より重度の表現型を示すことが多く、全例が成長障害を呈する。成長障害は出生前には軽度で、出生後は中等度ないし重度となり、吸啜/摂食障害を伴う。筋緊張低下やてんかん発作を伴う中枢神経系の構造的異常が所見として多くみられ、これに重度の発達遅滞/知的障害が加わる。罹患者は、言葉を発することができないことが多い。典型的骨格所見としては、第5指趾の低形成、他の指趾の爪の低形成、目立つ末節骨、脊柱側彎がある。罹患者の中には独歩が可能な例もみられる。消化器系の合併症、ヘルニアとともに、心血管系・泌尿生殖器系の合併症が多くみられる。
SMARCE1
SMARCE1の病的バリアントを有する罹患者は、重度の知的障害、典型的顔貌、第5指趾の爪の欠損と他の指趾の低形成を示す傾向にある。手は、細く長い指を特徴とする。罹患者は、在胎週数に比して小さいことが多く、出生後は低身長と重度の小頭症、複雑心奇形、摂食障害、てんかん発作を呈する。
SOX4
重症例は、筋緊張低下、痙性四肢不全麻痺、てんかんなどの神経学的症候を呈することがある。
SOX11
器官系や体に現れる異常よりも、神経発達上の異常がより多くみられる傾向にある。
浸透率
Coffin-Siris症候群の浸透率は100%と思われる。
2001年以前に報告されたものでいうと、CSSは男性よりも女性のほうが多い[Fleckら2001]ものの、分子レベルでの確認が行われた症例についていうと、男女比はほぼ均等である[Koshoら2014b,Santenら2014]。X連鎖顕性、性限定遺伝、ミトコンドリア遺伝をうかがわせるデータはみられない。
発生頻度
分子レベルでの確認がなされたCoffin-Siris症候群の既報罹患者は200症例に満たないことから、CSSの診断は稀にしか下されていないのが現状である。しかし、これは実際より過少に評価されているように思われ、罹患者の中には医療者側の注意を惹くに至っていないものがあるように思われる。
さらに言うと、知的障害の大規模コホートにおいて、その何人かにARID1Bの病的バリアントが同定されている[Hoyerら2012]ことから推測して、CSS関連遺伝子(ならびに、ごく軽度のCSS関連表現型として現れると思われる遺伝子)の病的バリアントの出現頻度は、知的障害を伴う症例について現在言われている数字より大きい可能性がある。
遺伝学的に関連のある疾患(同一アレル疾患)
ARID1A、ARID2、DPF2、SMARCC2、SMARCE1、SOX4、SOX11の病的バリアントについては、本GeneReviewで述べたもの以外の表現型はみられない。
ARID1B、SMARCA4、SMARCB1の病的バリアントについては、典型的なCoffin-Siris症候群の表現型以外の表現型が現れることがある。
- ARID1B
知的障害(ID)単独の罹患者数人に、ARID1B内の病的バリアントや、ARID1Bを含む領域の微小欠失がみられたとの報告がある[Nagamaniら2009,Hoyerら2012,Michelsonら2012]。知的障害に加え、脳梁無形成(ACC)、てんかん発作、多毛症、難聴、近視を呈する数人に、ARID1Bの欠失がみられたとの報告がみられる[Santenら2014]。「ARID1B関連疾患」のGeneReviewを参照されたい。
- SMARCA4
SMARCA4の生殖細胞系列の病的バリアントのヘテロ接合が、ラブドイド腫瘍発症素因になることが報告されている[Schneppenheimら2010,Hasselblattら2011,Biegelら2014]。
- SMARCB1
Kleefstra症候群の1罹患者で、de novoのp.Arg37Hisバリアントのヘテロ接合が同定されている[Kleefstraら2012]。これは当該タンパク質のN末端のバリアントであるが、CSSにおけるSMARCB1のバリアントは、SNF5ドメインのC末端で確認されている(「分子レベルの病原」の項を参照)。
SMARCB1の生殖細胞系列のヘテロ接合性病的バリアントが、腫瘍発生に関する古典的2ヒット説に従った形でラブドイド腫瘍好発症候群を惹起するとの報告がみられる[Roberts & Biegel 2009,Biegelら2014]。
SMARCB1の生殖細胞系列のヘテロ接合性病的バリアントはまた、多発性神経鞘腫(schwannomatosis)の原因ともなる。
多発性神経鞘腫は、浸透率の低い常染色体顕性の腫瘍抑制遺伝子症候群で、神経鞘腫の多発傾向や、(これより少ないながら)髄膜腫の好発傾向を特徴とする[Merkerら2012]。
当初、CSSと診断された罹患者で、その後、SMARCA2[Santenら2012,Tsurusakiら2012]ないしPHF6[Wieczorekら2013]の病的バリアントが発見されるに至った例が何例かあることは、注目に値する。これをさらによく調べてみた結果、これらの罹患者はそれぞれ、Nicolaides-Baraitser症候群、Borjeson-Forssman-Lehmann症候群のほうによりよく合致することが示唆されるに至った[Koshoら2014b,Tsurusakiら2014b,Zweierら2014]。
表1の脚注8と13、ならびに「鑑別診断」の項を参照されたい。
鑑別診断
Nicolaides-Baraitser症候群(NCBRS)
Nicolaides-Baraitser症候群は、疎な頭髪、皮下脂肪の減少に起因する指節関節や末節骨の隆起、特徴的な粗野な顔、小頭症、てんかん発作、発達遅滞/知的障害を特徴とする。発達遅滞/知的障害の程度は、NCBRSの半数近くで重度、3分の1で中等度、そして残りが軽度である。3分の1近くではスピーチの発達が起こらない。NCBRSでSMARCA2の病的バリアントのヘテロ接合の存在が同定された[Van Houdtら2012]後、当初、CSSであると考えられていた1罹患者を改めて評価し直したところ、その所見がNCBRSのほうによりよく一致することがわかった[Tsurusakiら2012]という事実は、注目に値する。NCBRSの遺伝形式は常染色体顕性遺伝である。現在わかっている罹患者は、すべてSMARCA2に生じたde novoの病的バリアントである。
Borjeson-Forssman-Lehmann症候群(BFLS)(OMIM 301900)
Borjeson-Forssman-Lehmann症候群は、男性の典型例については、重度の知的障害、てんかん、性腺機能低下、代謝低下、顕著な肥満、顔の皮下組織の腫脹、小さな眼瞼裂、大きいながら変形のない耳介を特徴とする。BFLSを引き起こすPHF6の病的バリアントを有する女性については、表現型の上でCSSといくぶんかの重なりがあることがわかっている[Wieczorekら2013]。ただそれでも、BFLSとCSSは明確に別の疾患であると考えられている[Zweierら2013]。
9トリソミーモザイク
9トリソミーモザイクを有する1症例で、顔面所見(広く球状の鼻)、男性型多毛症、第5指趾低形成といった点でCSS類似の所見がみられたとの報告がある[Kushnick & Adessa 1976]。
低身長-爪異形成-指趾骨異常症候群(Brachymorphism-onychodysplasia-dysphalangism[BOD]syndrome)(OMIM 113477)
BOD症候群は、低身長、小さく異形成の爪、第5指の短指、広い口と鼻、軽度の知的障害を特徴とする[Verloesら1993,Elliott & Teebi 2000]。最後の軽度の知的障害という点が、BOD症候群とCSSを鑑別する鍵になると思われる。というのは、CSSにおける認知障害の程度は、ほぼ常に中等度ないし重度だからである。遺伝形式は常染色体顕性遺伝であるように思われる。
DOORS(deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures)症候群
CSSとの共通所見としては、指趾末節骨低形成や爪の奇形、聴力喪失、神経学的異常がある。DOORS症候群は常染色体潜性遺伝を示し、TBC1D24の両アレル性病的バリアントに起因して生じる(「TBC1D24関連疾患」のGeneReviewを参照)。
胎児性アルコール症スペクトラム(Fetal alcohol spectrum;FAS)
FASでは、小さな爪、出生前・出生後の発育遅延、顔の形態異常、認知機能障害がみられることがある。
胎児ヒダントイン/フェニトイン胎芽症
この症候群は出生前のフェニトインへの曝露に起因して生じるもので、末節骨低形成を伴う小さな爪、顔の形態異常、三関節拇指、毛髪線低位、翼状短頸、発育遅延、認知機能障害が報告されている。
Mabry症候群(精神発達遅滞を伴う高ホスファターゼ症)(訳注:「2019年版骨系統疾患国際分類の和訳」では「精神遅滞、末節骨短縮および特徴的な顔貌を伴う高ホスファターゼ症」と訳されている)(OMIM 239300)
Mabry症候群は、発達遅滞、てんかん発作、粗野な顔、第5指趾低形成、血清アルカリホスファターゼ濃度の上昇を特徴とする[Gomes & Hunter 1970,Kruseら1988,Thompsonら2010]。遺伝形式は常染色体潜性遺伝で、PIGVの両アレル性病的バリアントに起因して生じる[Krawitzら2010]。
Cornelia de Lange症候群(CdLS)
典型的なCdLSは、特徴的頭蓋顔面所見(アーチ状の眉、眉毛癒合、上向きの鼻、小さな歯、小頭症)、発育遅延、四肢奇形を特徴とする。四肢奇形として、時にCSS類似の第5指低形成が現れることがある。他の所見としては、心奇形、消化器系の奇形、尿性器奇形がある。病的バリアントが現れる原因遺伝子は、NIPBL、SMC1A、SMC3、HDAC8、RAD21である。CdLSの遺伝形式は、常染色体顕性(NIPBL、SMC3、RAD21)もしくはX連鎖性(SMC1A、HDAC8)である。
4q欠失症候群
4q欠失症候群では、第5指趾の特徴的な彎曲した手掌/足底爪がみられ、これにより時に末節骨低形成様外観となる。
BICRA関連疾患(OMIM 619325)
BICRA関連疾患は、古典的なものでは、小頭症、前額部の突出、内眼角贅皮、目立つ鼻尖、耳介低位などを伴う粗野な顔を特徴とする。こうした症候は、CSS罹患者においてもよくみられる。Barishら[2020]は、BICRAの病的バリアントを有する14人の罹患者を同定し、その全員に、粗野な顔と種々の程度の知的障害がみられたことを報告している。BICRA関連疾患は、CSSとの間に表現型の類似性がみられるものの、これが臨床的にCSSと同類の疾患か、それとも明確に別の疾患かという点を確認するためには、BICRAの病的バリアントを有する罹患者の、より大規模なコホート研究が必要であろう。BICRA関連疾患は常染色体顕性の遺伝形式をとる。
SMARCD1関連疾患(OMIM 618779)
Nixonら[2019]は、SMARCD1の病的バリアントを有する5例を同定している。これらの罹患者には、発達遅滞、知的障害、筋緊張低下、摂食障害がみられた。SMARCD1関連疾患は、CSSとの間に表現型の類似性がみられるものの、これが臨床的にCSSと同類の疾患か、それとも明確に別の疾患かという点を確認するためには、SMARCD1の病的バリアントを有する罹患者の、より大規模なコホート研究が必要であろう。
臨床的マネジメント
最初の診断に続いて行う評価
Coffin-Siris症候群(CSS)と診断された罹患者については、疾患の範囲やニーズを把握するため、以下の評価を行うことが推奨される。
- 臨床遺伝医や遺伝カウンセラーとの面談
- 発達指標を記録し、神経症状ないし神経障害を発見するための神経学的検査ないし発達検査
- 必要に応じ、作業療法、言語治療、理学療法関連の評価
- 摂食障害や発育障害を念頭に行う消化器系の評価
- 必要に応じ、栄養士による栄養評価
- 散瞳眼底検査、視力検査を含む眼科的検査
- 難聴の評価を目的とした聴性脳幹反応検査や耳音響放射検査などの聴覚評価
- 構造的心奇形を評価するための心エコー
- 構造的腎奇形や尿性器奇形を評価するための腎超音波検査
症候に対する治療
- 最良の発達を誘導するための作業療法、理学療法、言語治療
- 摂食治療、栄養補助療法、ないし栄養学的要求を満たす上で必要であれば胃瘻造設・チューブ留置
- 屈折異常の補正に必要であれば眼鏡の使用、ならびに斜視や眼瞼下垂に対しては必要に応じ外科手術
- 必要に応じ補聴器の使用
二次的合併症の予防
二次的合併症の予防につながる治療や対策は、罹患者それぞれに推奨される治療と表裏一体のものである。それが発達治療であるかもしれないし、心臓・消化器・神経に関する適切な評価や治療であったり、眼科的、聴覚的サーベイランスであったりということもありうる。
定期的追跡評価
追跡評価は以下の形で行う。
- 発達状況や、治療的・教育的介入の成果をみるための、発達小児科医による年に1度の評価
- 必要に応じて摂食や体重増加をモニターしていくための、消化器医や摂食の専門家による年に1度のフォローアップ
- 眼科的、聴覚的異常に関する通常のフォローアップ
CSSでは腫瘍の発生は稀であるため、腫瘍に関するサーベイランスは有益とはされていない。
リスクを有する血縁者の評価
リスクを有する血縁者に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
妊娠に関する管理
CSS罹患者の女性の出産例は報告されていないので、妊娠に伴って生じうる合併症については不明である。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「Clinical Trials.gov」、ならびにヨーロッパの「EU Clinical Trials Register」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
Coffin-Siris症候群(CSS)は常染色体顕性の遺伝形式をとる。ただ、今日までに報告された罹患者の大多数は、de novoの病的バリアントの例である。
家族構成員のリスク
発端者の両親
- 現在までに報告されている発端者の大多数は、CSSを引き起こす遺伝子に生じたde novoの病的バリアントに起因するものである。
- De novoの病的バリアントに起因する罹患者の割合はわかっていないが、親が罹患者であったとする報告がほとんどみられないことから考えて、おそらく100%近いのではないかと思われる。
- 見かけ上、発端者の病的バリアントがde novoのものと思われる場合は、両親に対し、発端者で同定された病的バリアントに関する検査を行うことが推奨される。
- 発端者で同定された病的バリアントが、両親いずれの白血球DNAから検出されなかった場合は、発端者のもつ病的バリアントはde novoのものである可能性が濃厚である。
ただ、稀な例として、親が生殖細胞系列モザイクであったという可能性も考えられる[Ben-Salemら2016]。
- 親を評価してみると、実際は片親が罹患者であったにもかかわらず、表現型が軽微であったため、それまで検査をすり抜けてきたといったことがありうる。したがって、見かけ上、家族歴が陰性であるように思われても、適切な形で評価を行うまでは、家族歴陰性の確定はできない。
発端者の同胞
- 発端者の同胞の有するリスクは、発端者の両親の遺伝学的状態によって変わってくる。
- 発端者の片親も罹患者で、CSSを引き起こす遺伝子の病的バリアントをヘテロで有するという稀な状況の場合、同胞の有するリスクは50%である。
- 両親が臨床的にみて非罹患者ということであれば、同胞の有するリスクは低いものと思われる。
- 発端者で同定された病的バリアントが両親いずれの白血球DNAからも検出されない場合、同胞の有する再発リスクは、経験的に1%程度と思われる。それは、理論上、親の生殖細胞系列モザイクの可能性が残るからである。
- CSSの2姉妹例で、その父親が一部の症候を有していたとする1報告が存在する[Haspeslaghら1984]。これは親の体細胞(ならびに生殖細胞系列)モザイクを示唆するものと思われる。ただ、この例については分子レベルでの診断確定が行われておらず、罹患者の疾患がCSS以外のものであった可能性も考えられる。
発端者の子
CSS罹患者の子がCSS関連の病的バリアントを継承する可能性は50%である。
他の家族構成員
他の血縁者の有するリスクは、発端者の両親の状態によって変わってくる。罹患者の片親も罹患者であったという稀な例であれば、その血族にあたる人もリスクを有することになる。
関連する遺伝カウンセリング上の諸事項
家族計画
- 遺伝学的リスクの確定、出生前/着床前の遺伝学的検査を受けるかどうかの話し合いといったことに最も適しているのは、妊娠前の時期である。
- すでに罹患児をもつカップルに対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法であるとか、遺伝子・アレルバリアント・疾患等に対するわれわれの理解は、将来、より進歩していくことが予想される。そのため、分子診断の確定していない(すなわち、原因となった遺伝学的変化が未解明の)発端者のDNAについては、保存しておくことを検討すべきである。
出生前検査ならびに着床前の遺伝子検査
家系内の発端者の有する病的バリアントは、de novoのものである可能性が高いと考えられることから、その後の妊娠に際してのリスクは低いものと思われる。ただ、片親が生殖細胞系列モザイクを有する可能性があることから、一般集団よりは高リスクとなることが考えられる。そのため、出生前検査や着床前遺伝学的検査も1つのオプションとして検討に値する。
出生前検査の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。現在、多くの医療機関では、出生前検査を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- Coffin-Siris Syndrome Foundation
11611 106th Avenue NE
Kirkland WA 98034
Email: coffinsiris@gmail.com
www.coffinsiris.org
- Genetic and Rare Diseases Information Center (GARD)
P.O. Box 8126
Gaithersburg MD 20898-8126
Phone: 888-205-2311; 888-205-3223 (TTY); 301-251-4925
Fax: 301-251-4911
Email: GARDinfo@nih.gov
Coffin-Siris Syndrome
- American Association on Intellectual and Developmental Disabilities (AAIDD
)8403 Colesville Road
Suite 900
Silver Spring MD 20910
Phone: 202-387-1968
Fax: 202-387-2193
www.aaidd.org
- CDC Child Development
Phone: 800-CDC-INFO
Email: cdcinfo@cdc.gov
Intellectual Disability
- MedlinePlus
Intellectual Disability
- Clinical Registry of Individuals with Coffin-Siris Syndrome and Other BAF-Related Phenotypes
Email: Samantha.vergano@chkd.org
- CoRDS Registry
Sanford Research
2301 East 60th Street North
Sioux Falls SD 57104
Phone: 605-312-6423
Email: sanfordresearch@sanfordhealth.org
CoRDS Registry
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:Coffin-Siris症候群の遺伝子とデータベース
データは、以下の標準資料から作成したものである。遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:Coffin-Siris症候群関連のOMIMエントリー(内容の閲覧はOMIMへ)
| 35900 | COFFIN-SIRIS SYNDROME 1; CSS1 |
| 184430 | SRY-BOX 4; SOX4 |
| 600898 | SRY-BOX 11; SOX11 |
| 601607 | SWI/SNF-RELATED, MATRIX-ASSOCIATED, ACTIN-DEPENDENT REGULATOR OF CHROMATIN, SUBFAMILY B, MEMBER 1; SMARCB1 |
| 601671 | D4, ZINC, AND DOUBLE PHD FINGERS FAMILY, MEMBER 2; DPF2 |
| 601734 | SWI/SNF-RELATED, MATRIX-ASSOCIATED, ACTIN-DEPENDENT REGULATOR OF CHROMATIN, SUBFAMILY C, MEMBER 2; SMARCC2 |
| 603024 | AT-RICH INTERACTION DOMAIN-CONTAINING PROTEIN 1A; ARID1A |
| 603111 | SWI/SNF-RELATED, MATRIX-ASSOCIATED, ACTIN-DEPENDENT REGULATOR OF CHROMATIN, SUBFAMILY E, MEMBER 1; SMARCE1 |
| 603254 | SWI/SNF-RELATED, MATRIX-ASSOCIATED, ACTIN-DEPENDENT REGULATOR OF CHROMATIN, SUBFAMILY A, MEMBER 4; SMARCA4 |
| 609539 | AT-RICH INTERACTION DOMAIN-CONTAINING PROTEIN 2; ARID2 |
| 614556 | AT-RICH INTERACTION DOMAIN-CONTAINING PROTEIN 1B; ARID1B |
| 617808 | COFFIN-SIRIS SYNDROME 6; CSS6 |
| 618027 | COFFIN-SIRIS SYNDROME 7; CSS7 |
| 618362 | COFFIN-SIRIS SYNDROME 8; CSS8 |
| 618506 | INTELLECTUAL DEVELOPMENTAL DISORDER WITH SPEECH DELAY AND DYSMORPHIC FACIES; IDDSDF |
分子レベルの病原
これまでにCSSで同定されたタンパク質の多くは、最初に酵母やショウジョウバエのBRG1-and BRM-associated factor(BAF)複合体において発見されたタンパク質のヒトにおけるホモログである。BAF複合体は、もともとはmammalian switch/sucrose non-fermentable(mSWI/SNF)様ヌクレオソームリモデリング複合体と呼ばれていたものである。この複合体は、DNA刺激性のATPase活性を内包しており、これによりATP依存性にヒストン-DNAの結合を緩める働きをする[Ronanら2013]。SOX11は、神経発生、ならびに出生後のグリア細胞からニューロンへの転換過程において、BAF複合体の下流で作用するものと予想されている[Nincovicら2013]。
表2:Coffin-Siris症候群:疾患の発症メカニズム
| 遺伝子1 | メカニズム |
|---|---|
| ARID1A | 機能喪失型 |
| ARID1B | 機能喪失型 |
| ARID2 | 機能喪失型 |
| DPF2 | 機能喪失型 |
| PHF6 | |
| SMARCA4 | ドミナントネガティブ、もしくは異常機能獲得型 |
| SMARCB1 | ドミナントネガティブ、もしくは異常機能獲得型 |
| SMARCC2 | 機能喪失型 |
| SMARCE1 | ドミナントネガティブ、もしくは異常機能獲得型 |
| SOX4 | 機能喪失型 |
| SOX11 | 機能喪失型 |
- 表1の遺伝子をアルファベット順に示した。
表3:Coffin-Siris症候群:遺伝子特異的な検査技術の考慮事項
| 遺伝子1 | 特記事項 |
|---|---|
| ARID1A | 多くの病的バリアントがモザイクであるように思われる。 配列データを解析するにあたっては、このことを頭に入れておく必要がある。 |
| ARID1B | 異なるアイソフォームをコードする選択的スプライシングを受けた複数の転写バリアントが報告されている。 |
| SMARCA4 | この遺伝子については、異なるアイソフォームをコードする複数の転写バリアントが確認されている。 |
| SOX11 | SOX11の病的バリアントには、遺伝子の部分欠失や全欠失、HMG-box DNA-binding domainに生じたde novoのミスセンスバリアントなどがある3。 |
- 表1の遺伝子をアルファベット順に示した。
- Gazdaghら[2019](訳注:「2」の上付きの数字が表中に見当たらない。この論文はARID2に関するものなので、この表中に該当するものはないと思われる。)
- Tsurusakiら[2014a],Hempelら[2016]
表3:Coffin-Siris症候群:遺伝子ごとにみた注目すべき病的バリアント
| 遺伝子1 | 参照配列 | DNAヌクレオチドの変化 | 予想されるタンパク質の変化 | コメント[参考文献] |
|---|---|---|---|---|
| SMARCB1 | NM_003073.5 NP_003064.2 |
c.1085AGA[2] | p.Lys364del | De novoの反復性病的バリアント;罹患者は驚くほどよく似た臨床症候を示す2。 |
| SMARCC2 | NM_001330288.2 | c.1926+1G>T | スプライス部位変異 | De novoの反復性病的バリアント;罹患者は発達遅滞、スピーチの発達なしもしくは最小、筋緊張低下を示す3。 |
上記の変異は報告者の記載をそのまま載せたもので、GeneReviewsのスタッフが独自に変異の分類を検証したものではない。
GeneReviewsは、Human Genome Variation Society(varnomen.hgvs.org)の標準命名規則に準拠している。
命名規則の説明については、Quick Referenceを参照のこと。
- 表1の遺伝子をアルファベット順に示した。
- Koshoら[2014b]
- Macholら[2019]
癌ならびに良性腫瘍
SMRCA4
SMARCA4の生殖細胞病的バリアントのヘテロ接合により、ラブドイド腫瘍の罹患性が高まるとの報告がみられる。同様に、非定形類奇形腫やラブドイド腫瘍において、SMARCA4の体細胞病的バリアントもみられることが報告されている[Schneppenheimら2010,Hasselblattら2011,Biegelら2014]。
SMRCB1
生殖細胞系列におけるSMARCB1の病的バリアントのヘテロ接合が、ラブドイド腫瘍好発症候群を引き起こすとの報告がみられる。ラブドイド腫瘍好発症候群においては、腫瘍の大半において両アレル性の機能喪失型バリアントが生じており、これに呼応する形で、非定型類奇形腫やラブドイド腫瘍内でSMARCB1の体細胞性病的バリアントがみられる[Roberts & Biegel 2009,Biegelら2014]。
更新履歴:
- GeneReviews著者: Samantha Schrier Vergano, MD, Gijs Santen, MD, PhD, Dagmar Wieczorek, MD, Bernd Wollnik, MD, Naomichi Matsumoto, MD, PhD, and Matthew A Deardorff, MD, PhD
日本語訳者:仲 麻微、升野光雄、山内泰子、黒木良和(川崎医療福祉大学大学院 医療福祉学研究科 遺伝カウンセリングコース)
GeneReviews最終更新日: 2018.2.8. 日本語訳最終更新日: 2021.12.5. -
Gene Reviews著者: Samantha Schrier Vergano, MD, Gijs Santen, MD, PhD, Dagmar Wieczorek, MD, Bernd Wollnik, MD, Naomichi Matsumoto, MD, PhD, and Matthew A Deardorff, MD, PhD.
日本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日:2021.8.12. 日本語訳最終更新日: 2023.12.15.[in present]
![]()