頭蓋外胚葉異形成症
(Cranioectodermal Dysplasia)
[Synonyms:Sensenbrenner Syndrome]
Gene Reviews著者: Philip J Ostrowski, MD and Katrina Tatton-Brown, MD.
日本語訳者: 佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2022.12.15. 日本語訳最終更新日: 22023.3.27.
原文: Cranioectodermal Dysplasia
要約
疾患の特徴
頭蓋外胚葉異形成症(CED)は、骨格異常(狭い胸郭,近位肢の短縮,合指趾,多指趾,短指趾)、外胚葉症候(歯の低形成を伴う空隙歯列,無歯症,疎な毛髪,皮膚弛緩,爪の異常)、関節弛緩、発育不全、特徴的顔貌(前額部の突出,低位で単純な形の耳,縦に広い額,内眼角開離,内眼角贅皮,膨らんだ頰,下口唇反転)を伴う繊毛病の1つである。大多数の患児はネフロン癆を発症し、しばしば乳幼児期・小児期に末期腎不全に至る。そして、これが罹病や死亡の大きな原因になっている。肝線維症や網膜ジストロフィーがみられることもある。矢状縫合の早期癒合症に続発することの多い長頭症が、CEDと他の繊毛病を見分ける重要症候である。脳奇形や発達遅滞が生じることもある。
診断・検査
発端者におけるCEDの診断は、特徴的臨床症候・X線写真症候(高頻度にみられる症候2つに加えて、その他の異常を2つ、そのうち少なくとも1つは外胚葉症候―すなわち歯・毛髪・爪の異常であることが必要)を有すること、ないし、現時点でCEDの原因遺伝子であることがわかっている6つの遺伝子(IFT43,IFT52,IFT122,IFT140,WDR19,WDR35)の1つに両アレル性の病的バリアントが同定されることをもって確定する。
臨床的マネジメント
症状に対する治療:
必要に応じ、矢状縫合の早期癒合症を改善する手術を行う。これは、通常、満1歳未満の段階で行われる。また、多指趾についても外科的改善が必要になることがある。股関節形成不全に対しては整形外科的ケアを行う。標準治療基準を満たす例については、ヒト成長ホルモン治療が検討対象になりうる。歯の異常、ネフロン癆、肝臓病、心奇形、鼠径/臍ヘルニアに関しては標準治療を行う。進行性の視覚障害を有する例については、ロービジョン補助具、ならびに適切な教育プログラムを使用する。肺形成不全を有する新生児については、機械換気が必要になる場合がある。発達遅滞を伴う例については、言語治療、理学療法、ならびに適切な教育プログラムを行う。
定期的追跡評価:
乳児期から小児期にかけては、以下のような形で行う。
- 歯列の発達状況のモニタリング。
- 腎臓専門医の推奨に従って、早朝尿浸透圧試験、多尿に関する採尿検査、血圧・血清クレアチニン・血中尿素の評価、腎超音波検査。
- 肝臓専門医の推奨に従って、肝トランスアミナーゼ、肝臓の合成機能の評価。
- 網膜変性の初期の徴候を検知することを目的として行う4歳以降、年に1度の眼科的検査。
- 心臓病専門医による心臓検査、心電図、心エコー。
- プライマリケアへの来院ごとに行う発達の進行状況のチェック。遅延がみられた場合は、神経心理学的検査による正式な評価。
遺伝カウンセリング
CEDは常染色体潜性の遺伝形式をとる。CEDを引き起こす病的バリアントに関して、両親ともヘテロ接合者であることがわかっている場合、罹患者の同胞は、受胎の段階で、罹患者である可能性が25%、無症状の保因者である可能性が50%、罹患者でも保因者でもない可能性が25%である。家系内に存在するCEDを引き起こす病的バリアントが同定されている場合は、リスクを有する血族に対する保因者の検査、高リスクの妊娠に備えた出生前診断、着床前遺伝学的検査を行うことが可能である。第2三半期に超音波検査を行うことで、腎嚢胞、四肢短縮、多指趾といったものが確認されることがある。
診断
本疾患を示唆する所見
以下のような臨床所見・X線写真所見を示す例については、頭蓋外胚葉異形成症(CED)を疑う必要がある。
高頻度にみられる症候(75%超)
- 特徴的顔貌(例えば、前額部の突出,低位で単純な形の耳,高い額,内眼角開離,内眼角贅皮,膨らんだ頰,下口唇反転)
- 短指趾
- 長頭ならびに矢状縫合の早期癒合症
- 近位骨(多いのは上腕骨)の短縮(ならびに彎曲)
- 低身長
多くみられる症候(50%-75%)
- 狭い胸郭(肋骨形成不全と漏斗胸を伴う)
- 歯の異常(形態異常,空隙歯列,無歯症)
- 疎ないし薄い毛髪
- ネフロン癆(腎嚢胞,腎瘢痕化,超音波での高エコー輝度腎,尿細管間質性腎炎,腎機能/腎濃縮能の低下といったものを含む進行性腎疾患の1表現型)
- 発達遅滞(主として運動発達への影響)
比較的低頻度にみられる症候(25%-50%)
- 関節弛緩
- 肝臓病(肝線維症,肝硬変,肝腫大)
- 合指趾
- 多指趾
- 爪の異常
- 皮膚弛緩
- 反復性肺感染症
- 両側性鼠径ヘルニア
時としてみられる症候(25%未満)
- 網膜ジストロフィー
- 股関節形成不全
- 嚢胞性ヒグローマ
- 先天性心疾患
- 知的障害
注:本疾患を示唆する所見の一部(例えば、発達遅滞,歯の異常,網膜・腎・肝の異常)は、新生児期における評価の際にはみられないことがある。
診断の確定
エビデンスに基づく正式な診断基準は作成されていないものの、発端者におけるCEDの診断は、臨床的診断基準[Linら2013]をもとに、あるいは、本疾患を示唆する所見を有することに加え、分子遺伝学的検査にて表1に示した遺伝子の1つに両アレル性の病的バリアント(pathogenicとlikely pathogenicの両方を含む)がみられことをもって確定させることが可能である
注:(1)アメリカ臨床遺伝ゲノム学会(ACMG)/分子病理協会(AMP)のバリアントの解釈に関するガイドラインによると、「pathogenic」のバリアントと「likely pathogenic」のバリアントとは臨床の場では同義であり、ともに診断に供しうるものであると同時に、臨床的な意思決定に使用しうるものとされている[Richardsら2015]。本セクションで「病的バリアント」と言うとき、それは、あらゆるlikely pathogenicのバリアントまでを包含するものと理解されたい。
(2)表1に挙げた遺伝子の1つに両アレル性の意義不明バリアント(あるいは、既知の病的バリアントが1つと、意義不明バリアントが1つ)が同定された場合、それは、本疾患の診断を確定するものでも否定するものでもない。
臨床診断
発端者におけるCEDの臨床診断は、「本疾患を示唆する所見」にある臨床症候・X線写真症候のうち、高頻度にみられる症候2つに加え、それ以外の異常を2つ有し、そのうちの少なくとも1つが外胚葉症候(すなわち、歯・毛髪・爪の異常)であることをもって確定が可能である。矢状縫合の早期癒合症は、他のほとんどの繊毛病ではみられないCED特有の所見である[Linら2013]。
分子診断
発端者におけるCEDの分子診断は、「本疾患を示唆する所見」にある症候を有することに加え、分子遺伝学的検査にて表1に挙げた遺伝子の1つに両アレル性の病的バリアントが同定されることをもって確定が可能である。
分子遺伝学的検査のアプローチとしては、表現型に合わせて、遺伝子標的型検査(マルチ遺伝子パネル)と網羅的ゲノム検査(エクソームシーケンシング,ゲノムシーケンシング)とを組み合わせるやり方が考えられる。
遺伝子標的型検査の場合は、臨床医の側で関与している遺伝子の目星をつけておく必要があるが、ゲノム検査の場合、その必要はない。「本疾患を示唆する所見」にある特徴的所見を呈する例については、遺伝子標的型検査(「方法1」参照)で診断がつく可能性が高いように思われる一方、表現型からだけでは、その他数多く存在する外胚葉形成不全を伴う遺伝性疾患や骨格異常を伴う遺伝性疾患と区別が難しい例については、ゲノム検査(「方法2」参照)で診断がつく可能性が高いように思われる。
方法1
現況の表現型と直接関係のない遺伝子の意義不明バリアントや病的バリアントの検出を抑えつつ、疾患の遺伝的原因の特定につながる可能性が高いのは、表1に挙げた遺伝子、ならびにその他の関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネルである。
注:
(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝学的検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
方法2
表現型からだけでは、その他数多く存在する外胚葉形成不全や骨格異常を伴う遺伝性疾患と区別がつきにくいような場合であれば、網羅的ゲノム検査(この場合、臨床医の側で疑わしい遺伝子の目星をつけておく必要はない)が最良の選択肢となる。エクソームシーケンシングが広く用いられているが、ゲノムシーケンシングを使用することも可能である。
網羅的ゲノム検査の基礎的情報についてはここをクリック。
ゲノム検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
表1:頭蓋外胚葉異形成症で用いられる分子遺伝学的検査
| 遺伝子1,2 | CEDの中でその遺伝子の病的バリアントの占める割合 | その手法で病的バリアント3が検出される割合 | |
|---|---|---|---|
| 配列解析4 | 遺伝子標的型欠失/重複解析5 | ||
| IFT43 | 2/71(3%) | 100%6 | 報告例なし7 |
| IFT52 | 1/71(1%) | 100%6 | 報告例なし7 |
| IFT122 | 13/71(18%) | 100%6 | 報告例なし7 |
| IFT140 | 3/71(4%) | 60%8,9 | 40%8 |
| WDR19 | 5/71(7%) | 100%6 | 報告例なし7 |
| WDR35 | 23/71(32%) | 100%6 | 報告例なし10 |
| 不明 | 24/71(34%) | 対象外 | |
- 遺伝子の配列はアルファベット順。
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- この遺伝子で検出されているバリアントの情報については、「分子遺伝学」の項を参照のこと。
- 配列解析を行うことで、benign、likely benign、意義不明、likely pathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、遺伝子内の小さな欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
- 遺伝子標的型欠失/重複解析では、遺伝子内の欠失や重複が検出される。具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失/重複の検出を目的に設計された遺伝子標的型マイクロアレイなど、さまざまなものがある。
- データは、ヒト遺伝子変異データベース(HGMD)の購読ベースの専門家向けデータ[Stensonら2020]を基にしたもの。
- これまでに遺伝子標的型欠失/重複解析で欠失/重複が検出された例はみられない。
- Walczak-Sztulpaら[2020]
- Bayatら[2017]
- 頭蓋外胚葉異形成症を引き起こすWDR35の欠失/重複の報告例はみられない。WDR35のエクソン欠失によって生じる表現型については、「遺伝子の上で関連のある疾患」の項を併せて参照されたい。
臨床的特徴
臨床像
頭蓋外胚葉異形成症(CED)は繊毛病の1つで、骨格、外胚葉(歯・毛髪・爪)、網膜、腎、肝、肺に大きな異常が生じ、時として脳にも影響が及ぶ疾患である。今のところ、詳細な報告例は少数にとどまっており、加えて、分子レベルでの確認が行われている例はさらに少ないという現実もあり、CEDの表現型に関し現時点でわかっていることはそれほど多くない。
表1に挙げた遺伝子の1つに両アレル性の病的バリアントを有する例の臨床報告は、現在までに44例存在する[Zaffanelloら2006,Fry ら2009,Gilissenら2010,Walczak-Sztulpaら2010,Artsら2011,Bredrupら2011,Bacinoら2012,Hofferら2013,Linら2013,Alazamiら2014,Tsurusakiら2014,Liら2015,Daoudら2016,Girishaら2016,Moosaら2016,Smithら2016,Antonyら2017,Bayatら2017,Silveiraら2017,Walczak-Sztulpaら2017,Xuら2017,Yoshikawaら2017,Ackahら2018,Córdova-Fletesら2018,Walczak-Sztulpaら2020,ならびに著者自身の未発表例]。
以下に述べる本疾患の表現型の特徴は、これらの報告に基づくものである。
表2:頭蓋外胚葉異形成症でみられる代表的症候
| 頻度 | 症候 |
|---|---|
| 高頻度にみられる症候 (75%超) |
特徴的顔貌(前額部の突出,低位で単純な形の耳,高い額,内眼角開離,内眼角贅皮,膨らんだ頰,下口唇反転) |
| 短指趾 | |
| 長頭,ならびに矢状縫合早期癒合症 | |
| 近位骨の短縮/彎曲(最も多いのは上腕骨) | |
| 低身長 | |
| 多くみられる症候 (50%-75%) |
肋骨形成不全と漏斗胸を伴う狭い胸郭 |
| 歯の異常(形態異常歯,空隙歯列,無歯症) | |
| 疎ないし薄い毛髪 | |
| ネフロン癆 | |
| 発達遅滞(最も多いのは運動発達遅滞) | |
| 比較的低頻度にみられる症候 (25%-50%) |
関節弛緩 |
| 肝疾患(肝線維症,肝硬変,肝腫大) | |
| 合指趾 | |
| 多指趾 | |
| 爪の異常 | |
| 皮膚弛緩 | |
| 反復性肺感染症 | |
| 両側性鼠径ヘルニア | |
| 時としてみられる症候 (25%未満) |
網膜ジストロフィー |
| 股関節形成不全 | |
| 嚢胞性ヒグローマ | |
| 先天性心疾患 | |
| 知的障害 |
特徴的顔貌
特徴的顔貌は出生時から認められ、CEDのほぼ全例でみられる(図1参照)。具体的には、前額部の突出、側頭骨間距離の短縮、縦に広い額、低位で単純形態で後方に傾いた耳、内眼角開離、内眼角贅皮、眼瞼裂斜下/斜上、膨らんだ頰、小下顎、下口唇反転、上向きの鼻孔などがある。
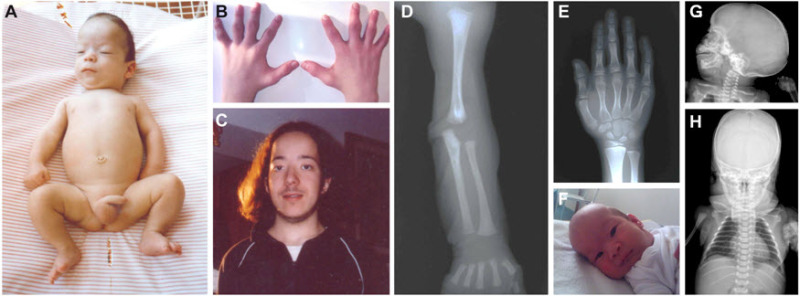
図1:頭蓋外胚葉異形成症の臨床症候とX線写真症候
症例1(A-E):
A.腕の近位肢節短縮、狭い胸郭、特徴的顔貌(前額部の突出,眼間開離,耳介低位)を伴うCED新生児。
B.指節間関節の肥大を伴う短く幅広の手(16歳時)。
C.若い成人期におけるCEDの顔貌の特徴。
D.近位肢節短縮を示す新生児期のX線写真
E.指節骨の短縮を示す9歳時のX線写真
症例2(F-H):
F.前額部の突出、両側性の内眼角贅皮などの特徴的顔貌を呈する女児。
G.新生児期における長頭症を示すX線写真
H.狭い胸郭を示すX線写真
患者やその家族の同意を得て画像を掲載。
長頭
長頭(頭部の幅径に対し、長径が明らかに大きい状態)、ならびに前額部の突出は、通常、矢状縫合の早期癒合に伴う続発性のもので、この矢状縫合の早期癒合は、出生の段階ですでに存在することが多い。矢状縫合の早期癒合に関しては、同胞対間で不一致である場合がある[Artsら2011,Bredrupら2011]。
骨格所見
- 手足
出生前の超音波検査で、妊娠中期に多指趾症が発見されることがある[Konstantinidouら2009]。新生児については、短指趾(形態異常を伴う中節骨・末節骨の短小化)、軸後性多指趾、皮膚性合指趾(最も多いのは第2-3趾の軽度の皮膚性合趾)がしばしばみられる。指趾節骨の骨端は、X線写真で異常がみられないこともあれば、扁平もしくは円錐形を示す場合もある[Fryら2009]。ばらつきの幅があるものの、手足にみられるその他の異常としては、第5指の彎指、手掌屈曲線の異常、手指の屈曲制限、骨粗鬆症、サンダルギャップ、3指節拇趾などがある[Gilissenら2010,Bacinoら2012,Hofferら2013,Linら2013]。
- 胸郭
全例にというわけではないものの、短く形成不全の肋骨を伴う狭い胸郭が、早ければ妊娠中期に認められることがある。ただ、もっと多いのは出生時に発見される例である[Linら2013]。漏斗胸もしばしばみられる[Hofferら2013]。肋骨の変形(例えば、短肋骨やハンガー形肋骨)については、小児期に正常化することがある[Bacinoら2012]。
- 近位長骨の短縮(ならびに彎曲)
近位長骨の短縮や彎曲は、早ければ妊娠23週に認められる。短縮は下肢より上肢のほうに顕著であることが多く、上腕骨に特に強く現れる。長骨は彎曲し、骨端が平坦化して、末広がりの骨幹端を示すことがある[Bacinoら2012,Hofferら2013,Linら2013]。
- 発育不全
出生時の身長は、胎齢比較で、正常範囲内にある例がみられる一方、3パーセンタイルを下回るような例もみられる。乳児についても、身長が3パーセンタイル未満の発育不全を示すことがあり、また、身長が5パーセンタイルから10パーセンタイルの間にあるような例もみられる[Bacinoら2013]。
外胚葉の問題
- 歯
歯の萌出は遅延することが多い。乳歯は概して小さく、空隙歯列となる。無歯症、エナメル質の欠損、タウロドント、癒合歯、円錐歯の報告もみられる。これと同様の特徴は永久歯においてもみられる。無歯症は上下どちらの歯列にも生じる[Fryら2009,Gilissenら2010,Walczak-Sztulpaら2010,Artsら2011,Bredrupら2011]。
- 皮膚
出生前の超音波検査で、妊娠中期に翼状頸や皮膚の肥厚が明らかになることがある。乳児期以降の全身性皮膚弛緩、ならびに余剰皮膚による襞形成が報告されている。皮膚の襞は、特に頸部、足首、手首の部分に多くみられる。皮膚は乾燥していることがあり、これまでに過角化の報告がみられる[Artsら2011,Bredrupら2011,Bacinoら2012]。
- 毛髪
CEDの乳幼児の毛髪は、多くの場合、疎で細い。毛髪は低色素で、直径も小さいことがある。毛髪は、成長も遅いことがある[Artsら2011,Linら2013]。ただ、中には小児期のうちに毛髪の成長が正常化するような例もみられる[Konstantinidouら2009]。
- 爪
爪は、乳児期以降、短く幅広で成長が遅いという特徴を示す[Hofferら2013]。
腎
腎疾患は、ネフロン癆(尿細管間質性腎炎)に起因して生じる。60%-70%を超えるCED罹患者において、腎不全が報告されている。中には、出生前の妊娠中期に、羊水過多/過少と小嚢胞腎の形で末期腎不全(ESKD)が現れる例もみられるものの、多くの場合は、幼児期(2歳以降)に腎臓病の最初の徴候が現れる[Bacinoら2012]。最初、腎の濃縮能低下から、多尿・多飲に至る。夜間遺尿がみられる場合もある。後には、腎不全や濾過障害の結果として、高血圧、タンパク尿、血尿、電解質平衡障害に至る疾患の経過をとる。小児の21人中10人で、腎臓病がESKDに進行していた。ただ、注意が必要なのは、これは限られた追跡期間中の数字であって、さらに期間が経過すれば、この数字はさらに上がっていく可能性があるという点である。大多数の小児は、2歳から6歳の間にESKDに至っていた[Hofferら2013,Linら2013]。
乳幼児期の腎超音波検査にて、腎は、正常サイズもしくは小さく、高エコー輝度と腎皮髄分化不良を呈することが多い[Linら2013]。腎生検では、炎症細胞浸潤を伴う間質線維化、尿細管萎縮、糸球体硬化症に加え、時に嚢胞が認められる[Obikaneら2006,Konstantinidouら2009,Bredrupら2011,Linら2013]。嚢胞は、疾患が進行した例でみられる。
肝
肝臓については、進行性肝疾患の徴候を伴わない肝脾腫から、(反復性)高ビリルビン血症や、新生児期の入院を要する胆汁鬱滞性疾患などの広範囲に及ぶ肝異常に至るまで、その所見は多様である[Walczak-Sztulpaら2010,Bacinoら2012]。乳児で、肝硬変、胆管増生を伴う重度の胆汁鬱滞、急性胆管炎の各例が報告されている[Zaffanelloら2006,Artsら2011,Bacinoら2012,Linら2013]。肝嚢胞については、3歳ならびに4歳で発見された例がみられる[Hofferら2013]が、わずか10ヵ月で確認された例も存在する[Linら2013]。
肝疾患に関する長期データは存在しないものの、肝線維症と肝硬変に関する長期予後は不良とみられる。
眼
眼所見としては、網膜ジストロフィーや眼振がある[Bredrupら2011,Linら2013]。
生後数年の段階で、夜盲がみられることも多い[Bredrupら2011]。暗所視・明所視網膜電図の異常が、早いものでは4歳から11歳で報告されており、眼底検査では、一部の罹患者に動脈の狭細化や骨小体様色素沈着が、早いもので5歳から11歳で確認されている[Bredrupら2011]。網膜ジストロフィーの自然経過に関する報告はなされていないものの、Bardet-Biedl症候群など症状の重なる繊毛病においては、夜盲が進行して、多くの場合、成人初期には法的盲(訳注:アメリカ政府が用いる行政上の定義で、矯正視力が0.1以下の状態をいう)に至る(「Bardet-Biedl症候群」のGeneReviewを参照)。CEDの予後についても、おそらくこれと同様であろうと思われる。
その他の眼科的所見としては、遠視、近視、内斜視、近視性/遠視性乱視、幅広の眼瞼(眼瞼の水平方向の長さが過剰な状態)などがある[Konstantinidouら2009,Bredrupら2011,Hofferら2013,Linら2013]。
肺
CEDの子どもは、乳幼児期に、肺形成不全や反復性呼吸器感染症に起因して、生命を脅かすような呼吸窮迫を経験する場合がある。喘息や気胸の報告もみられる[Bredrupら2011,Bacinoら2012,Hofferら2013]。出生後の呼吸窮迫や幼児期の肺炎がもとで死亡する子どもも多い[Tamaiら2002]。反復性呼吸器感染症の頻度は、時を経るに従って低下していくと報告されている[Konstantinidouら2009]。
心奇形
心奇形としては、動脈管開存、心房中隔欠損、心室中隔欠損などがある。僧帽弁・三尖弁の肥厚、心室肥大/拡大、末梢肺動脈狭窄の報告もみられる[Artsら2011,Bacinoら2012]。Bacinoら[2012]は、不整脈と心房中隔欠損が3歳で消退した罹患児の1例を報告している。
中枢神経系
大多数の子どもについては、中枢神経系の発達は正常であるが、一部、軽度の発達遅滞が報告されている[Hofferら2013,Linら2013]。一人座りは9-15ヵ月、歩行は3歳までずれ込むことがある[Fryら2009,Bacinoら2012,Hofferら2013]。スピーチの遅延は、19ヵ月の段階で数語といったものから、5歳で発語なしのものまで、幅がみられる[Hofferら2013]。罹患者に対する言語治療や理学療法の効果に関しては、情報が得られていない現状である。認知機能や社会技能はふつう正常であるものの、罹患者の中には知的障害の診断がなされる例も一部みられる[Fryら2009,Alazamiら2014,Liら2015]。
脳の画像診断では、皮質萎縮、脳室拡大、大槽の拡大、脳梁形成不全、局在性微小形成不全、脳外の液貯留腔の拡大、後頭蓋窩の大きな嚢胞、Dandy-Walker奇形などが明らかになっている[Zannolliら2001,Fryら2009,Konstantinidouら2009,Bacinoら2012,Hofferら2013,Linら2013,Girishaら2016,Walczak-Sztulpaら2020]。
その他
- 関節弛緩
関節弛緩が新生児期からみられることがある[Fryら2009]。
- ヘルニア
鼠径ヘルニア(両側性)ないし臍ヘルニアが、新生児期あるいは1歳未満の段階でみられることがある[Fryら2009,Walczak-Sztulpaら2010]。
寿命
CED患者の罹病率は高く、入院の頻度が高く、また入院期間が長くなりがちである[Bacinoら2012]。死亡率についてはよくわかっていないものの、CED罹患児65人中10人が、呼吸不全[Levinら1977,Savillら1997,Tamaiら2002]、心不全[Ekeら1996,Savillら1997,Bacinoら2012,Silveiraら2017,Walczak-Sztulpaら2017]、血液量減少性ショック(凝固障害に起因するもの)[Bacinoら2012]、ならびに不明な原因[Linら2013,Antonyら2017]により、7歳未満で死亡している。CED罹患者の大多数については経年的データが得られていないことを考慮すると、実際の死亡率の数字は、さらに高い可能性があろう。なお、少なくとも3人のCED罹患者が成人期まで生存していることが確認されている(Bredrupら[2011],ならびに図1を参照)。
遺伝子ごとにみた表現型との相関
既知の6つの原因遺伝子(すなわち、ITF43,ITF52,IFT122,IFT140,WDR19,WDR35)の両アレル性病的バリアントによって生じる表現型の、それぞれの違いを区別することはできない[Walczak-Sztulpaら2010,Artsら2011,Bredrupら2011,Bacinoら2012,Hofferら2013,Girishaら2016,Bayatら2017]。
遺伝型-表現型相関
遺伝型-表現型相関として確認されたものは存在しない。
疾患名について
CEDは最初に、長頭、近位肢節の骨の短縮、短指趾、外胚葉形成不全を伴う同胞対について、Sensenbrenner症候群という形で報告された[Sensenbrennerら1975]。続いて、Levinら[1977]が、これとは別の2家系の罹患例を報告し、その際、名を頭蓋外胚葉異形成症と改めた。
発生頻度
CEDは稀な疾患で、正確な発生頻度はわかっていない。これまでに報告された罹患者は100人未満である。
遺伝学的に関連のある疾患(同一アレル疾患)
IFT122の病的バリアントに起因して生じるものとしては、本GeneReviewで述べたもの以外の表現型は知られていない。
注:IFT122の両アレル性病的バリアントを有する1罹患者が、Beemer-Langer症候群(短肋骨多指症候群Ⅳ型)と診断された例が存在するものの、その後のレビューで、この例の表現型は頭蓋外胚葉異形成症(CED)のほうにより合致していることが判明している[Silveiraら2017]。
軽度の多発性嚢胞腎の表現型を有する例で、IFT140のヘテロ接合性病的バリアントが同定されている(「多発性嚢胞腎,常染色体顕性」のGeneReviewを参照)。
IFT43、IFT140の両アレル性病的バリアントについては、それぞれ、単発性網膜色素変性症81(OMIM 617871)、単発性網膜色素変性症80(OMIM 617781)を引き起こす。IFT43、IFT52、IFT140、WDR19、WDR35の生殖細胞系列病的バリアントに起因して生じるCED以外の常染色体潜性の表現型については、表3にまとめて示した。これらの疾患は、CEDと重なる症候を表現型として有していることから、「鑑別診断」の項においても検討していくこととする。
表3:頭蓋外胚葉異形成症との鑑別において頭に入れておくべき同一アレル疾患
| 遺伝子 | 疾患名 |
|---|---|
| IFT43 | 短肋骨多指症候群未分類1 |
| IFT52 | Jeune呼吸不全性胸郭異形成症1 |
| IFT140 | Jeune呼吸不全性胸郭異形成症1 |
| Mainzer-Saldino症候群1 | |
| Opitz三角頭蓋C症候群2 | |
| WDR19 | Jeune呼吸不全性胸郭異形成症1 |
| ネフロン癆 | |
| Senior-Løken症候群(OMIM 616307) | |
| WDR35 | 軟骨外胚葉性異形成症(Ellis-van Creveld症候群1 |
| 短肋骨多指症候群5型1 | |
| 短肋骨多指症候群未分類1 |
- Mortierら[2019]
- Peña-Padillaら[2017]
鑑別診断
頭蓋外胚葉異形成症(CED)は、細胞においてアンテナの働きをしているように見える小器官である繊毛の不具合によって引き起こされる、一連のスペクトラムを構成する疾患群の1つである(図2参照)[Huber & Cormier-Daire 2012]。こうした疾患群は、繊毛病と総称され、その表現型は互いに大きく重なり合っている。繊毛病の特徴的臨床症候としては、嚢胞性腎疾患、網膜ジストロフィー、肋骨・指節骨・長骨の短縮、多指趾、肝線維症、発達遅滞といったものがある。
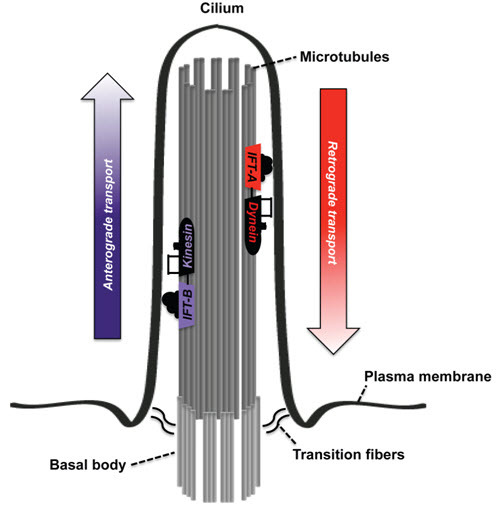
図2:繊毛の構造ならびに繊毛輸送の仕組みを示した模式図
繊毛とは、細胞の頂端膜から突き出た尾状の突起で、2つの部分から構成される。その1つは、基底小体(basal body)で、この部分から繊毛が組み上げられていく。そしてもう1つは、形質膜から突き出る軸糸である。繊毛の組み立て、ならびにシグナル伝達は、ともに、繊毛内輸送(intraflagellar transport;IFT)として知られる繊毛の輸送機能に依存する形で進んでいく。この輸送のプロセスは、軸糸の微小管に沿って基底部から先端部へ(順行性輸送)、そして反対方向へ(逆行性輸送)という双方向性に生じる。順行性輸送はキネシン2モーターとIFT-B複合体が推進する一方、逆行性輸送のほうはダイニン2モーターとIFT-A複合体が制御する。IFT-A複合体は6種のタンパク質から成り、頭蓋外胚葉異形成症罹患者においては、これらのIFT-A複合体タンパク質をコードする6つの遺伝子のうちの4つで、両アレル性病的バリアントが同定されている。IFT-B複合体は、少なくとも12種のタンパク質で構成されている。
繊毛病の中にあっては、Jeune呼吸不全性胸郭異形成症、Mainzer-Saldino症候群、Ellis-van Creveld症候群、短肋骨多指症候群が最もCEDに類似している(表4参照)。これらの常染色体潜性骨系統繊毛病は、OMIMにおいては、短肋骨胸郭異形成症と呼ばれている(OMIM PS208500)。
表4:頭蓋外胚葉異形成症との鑑別診断に関係してくる骨格性の繊毛病
| 遺伝子 | 疾患名 | 重なる症候 | 異なる症候 | |
|---|---|---|---|---|
| 骨格の異常 | 骨格以外の症候 | |||
| CEP120 DYNC2H1 DYN2LI1 IFT52 IFT80 IFT81 IFT140 IFT172 INTU KIAA0586 NEK1 TCTEX1D2 TTC21B WDR19 WDR34 WDR60 (OMIM PS208500) |
Jeune呼吸不全性胸郭異形成症(JATD) | 多指趾,短指趾,近位肢短縮 | 嚢胞性腎疾患,肝奇形,網膜ジストロフィー | JATDの表現型は、CEDと強く重なる。 狭い胸郭は両疾患でみられるものの、概してCEDは軽度である一方、JATDは顕著で、しばしば重度の呼吸窮迫に至る。 JATDの新生児・乳児10人のレビューで、うち6人が呼吸不全のため死亡している1。 |
| Mainzer-Saldino症候群(MZSDS) | 円錐形の指節骨骨端, ばらつきはあるものの、狭い胸郭や舟状頭蓋もみられる2。 |
網膜ジストロフィー,ネフロン癆 ばらつきはあるものの、小脳性運動失調や肝線維症もみられる2。 |
CEDでみられる特徴的外胚葉症候は、MZSDSでは通常みられない。 | |
| 短肋骨多指症候群(SRPS)1,3 | 極端に短い四肢や肋骨(重度の狭い胸郭),多指趾 | 種々の器官に現れる奇形4 | 重度の狭い胸郭により、SRPSは周産期致死性である。 | |
| EVC EVC2 WDR35 (OMIM 225500) |
Ellis-van Creveld (EVC)症候群3 | 軸後性多指趾,四肢・肋骨の短縮 | 毛髪・爪・歯に現れる外胚葉形成不全5,先天性心疾患(EVC症候群の主要所見:心房を中心とした中隔欠損) | 心疾患の出現頻度(罹患者の60%6)は、CEDより高い。 |
- Oberklaidら[1977]
- Perraultら[2012]
- SRPSとEVCについては、ここに挙げた遺伝子以外のWDR35の病的バリアントによって生じることもある(「遺伝子の上で関連のある疾患」の項を参照)。
- Elçioglu & Hall [2002],Huber & Cormier-Daire [2012]
- Huber & Cormier-Daire [2012]
- Ruiz-Pepezら[2000],Baujat & Le Merrer [2007]
CEDと臨床症候の重なるその他の繊毛病としては、単発性ネフロン癆、単発性網膜ジストロフィー、Caroli病、Senior-Løken症候群、Joubert症候群、Meckel-Gruber症候群(OMIM PS249000)、Bardet-Biedl症候群などがある[Huber & Cormier-Daire 2012]。
- Senior-Løken症候群(OMIM 266900)
Senior-Løken症候群は、ネフロン癆と網膜ジストロフィーを特徴とし、遺伝的異質性をもった常染色体潜性遺伝疾患である。Senior-Løken症候群では、これまでに、CEP290、IQCB1、NPHP1、NPHP4、SDCCAG8、TRAF3IP1、WDR19の病的バリアントが特定されている。(注:WDR19の病的バリアントはCEDの原因にもなっている。)
- Caroli病(OMIM 600643)
Caroli病は、多発性肝嚢胞症と胆管炎を特徴とする。これは、常染色体潜性多発性嚢胞腎スペクトラムの一部とされており、単発所見として現れることもあれば、多発性嚢胞腎疾患のその他の症候と組み合わさって現れることもある。
- 常染色体潜性網膜ジストロフィー
常染色体潜性網膜ジストロフィー(網膜色素変性症とも呼ばれる)は単発所見として現れることもあれば、CEDのような症候群性疾患に伴って現れることもある。網膜色素変性症は、ふつう、まず夜盲症の形で現れ、視細胞(杆体細胞・錐体細胞)が失われることで、後には完全失明に進行することもある。眼底は、しばしば網膜血管の狭細化を示し、周辺部に異常な色素沈着(骨小体様色素沈着と呼ばれる)が現れることがある。単発性網膜色素変性症家系の50%超は、常染色体潜性型である。網膜色素変性症の原因遺伝子は50を超える数が存在し、こうした遺伝子のほぼ3分の2が繊毛タンパク質をコードしている。
臨床的マネジメント
最初の診断に続いて行う評価
頭蓋外胚葉異形成症(CED)と診断された新生児あるいは乳児については、疾患の範囲やニーズを把握するため、診断に至る過程ですでに実施済でなければ、表5にまとめた評価を行うことが推奨される。
表5:頭蓋外胚葉異形成症罹患者の最初の診断後に行うことが推奨される評価
| 系/懸念事項 | 評価 | コメント |
|---|---|---|
| 矢状縫合早期癒合症 | 長頭を呈する例については、頭部CT検査 | 頭蓋縫合早期癒合症の評価を目的として行う。 |
| 骨格症候 | 胸郭や長骨のX線写真 | |
| 外胚葉症候 | 皮膚、毛髪、爪、歯の診査 | |
| 歯の異常 | 歯科的評価 | |
| ネフロン癆 |
|
異常が検出された場合は、腎生検が行われることが多い。 |
| 肝線維症 |
|
|
| 網膜ジストロフィー | 眼科的評価 | 視力低下がみられる場合は、4歳までに網膜電図と眼底検査を行う場合あり。 |
| 肺の症候(呼吸窮迫,喘息,気胸) | 呼吸器専門医による評価 | |
| 心奇形 | 心電図や心エコーなどの心評価 | |
| 発達遅滞 | 発達評価 | 遅滞を有する例については原因を評価するため、脳のMRIを行う。 |
| 遺伝カウンセリング | 遺伝の専門医療職1の手で行う。 | 医学的、個人的な意思決定の用に資するべく、本人や家族に対し、CEDの本質、遺伝形式、そのもつ意味についての情報提供を行う。 |
| 家族への支援/情報資源 | 以下の必要性に関する評価を行う。
|
- 臨床遺伝医、認定遺伝カウンセラー、認定上級遺伝看護師をいう。
症候に対する治療
表5:頭蓋外胚葉異形成症罹患者の症候に対する治療
| 症候/懸念事項 | 治療 | 考慮事項/その他 |
|---|---|---|
| 頭蓋縫合早期癒合症 | 通常は、満1歳に達する前に外科的治療 | |
| 多指趾 | 外科的改善を図るのも一手 | |
| 股関節形成不全 | 必要に応じ整形外科的ケア | |
| 低身長 | 成長ホルモン治療の基準を満たす場合に、これを行う。 | この治療が奏功すると思われる重度の発育不全を呈する子どもについてのみ行う。 |
| 歯の異常 | 歯科医師ないし口腔外科医による標準治療 | 歯の器質的異常ないし無歯症に対し適期に介入することで、審美的、機能的心理的問題の低減につながることあり。 |
| ネフロン癆 | 腎臓専門医による治療 | 段階の進行した例については、腎移植も選択肢となる。 |
| 肝線維症 | 肝臓専門医による治療 | 段階の進行した例については、肝移植も選択肢となる。 |
| 進行性視覚障害 | 眼科医によるロービジョン補助具,ならびに適切な教育プログラム | |
| 肺形成不全 | 罹患新生児については機械換気が必要になることあり。 | |
| 呼吸器感染症 | ・胸部X線写真、ならびに肺炎が疑われる場合は喀痰分析 ・必要に応じ抗生物質 |
反復性呼吸器感染症を起こしやすい例については、長期予防投薬が必要になる。 |
| 心奇形 | 心臓病専門医による治療 | |
| 発達遅滞 | ・言語治療ならびに理学療法 ・必要に応じ、早期介入ないし個別的教育計画(IEP) |
|
| 鼠径/臍ヘルニア | 外科的介入 |
定期的追跡評価
表7:頭蓋外胚葉異形成症罹患者で推奨される定期的追跡評価
| 系/懸念事項 | 評価 | 実施頻度 |
|---|---|---|
| 歯の異常 | 歯質のダメージと無歯症を調べるための歯科的検査 | 1歳から始め、6ヵ月ごと |
| ネフロン癆 |
|
診断後の臨床経過に従い、腎臓専門医の手で行う。 |
| 肝線維症 | トランスアミナーゼと肝合成機能の測定 | 診断後の臨床経過に従い、肝臓専門医の手で行う。 |
| 網膜ジストロフィー | 眼科的検査 | 4歳から始め、年に1度。 視力低下がみられる場合は、それより早く網膜電図や眼底検査を行うことあり。 |
| 心奇形 | 心臓病専門医の手で心臓検査、心電図、心エコーを行う。 | 最初の所見によっては、診断後の臨床経過に従い、また心臓病専門医の推奨に従ってこれを行う。 |
| 発達遅滞 | 発達評価 | ・乳幼児期、小児期に、来院ごとに行う。 ・遅滞がみられる場合は、神経心理士の手で正式な評価を行う。 |
避けるべき薬剤/環境
腎臓病がみられる場合は、腎臓専門医のほうから食餌中のカリウム、リンの制限が推奨される場合がある。
腎に問題がある例ついては、NSAIDのクラスに属する薬剤など、腎毒性を有する薬剤も相対的禁忌となる場合がある。罹患者は、必要に応じ腎臓専門医の管理下に置かれ、腎毒性があって避けるべき薬剤に関する説明を受ける必要がある。
リスクを有する血縁者の評価
家系内に存在するCEDを引き起こす病的バリアントが同定されている場合は、早期診断、ならびに呼吸器合併症、腎疾患、肝疾患、視覚障害を中心に、適切な管理と追跡評価ができるよう、リスクを有する乳児に対しては、その遺伝的状態を明らかにしておくことが望ましい。
家系内に存在する病的バリアントが未知の場合は、リスクを有する子どもに対して以下のことが推奨される。
- 新生児:小児科医による身体の診査、そして、臨床所見によっては臨床遺伝医との面談。
- 生後3ヵ月まで:超音波検査、ならびに血圧・血清クレアチニン濃度・肝酵素の測定をはじめとする腎評価と肝評価。
- 6ヵ月ごと:腎評価と肝評価を繰り返す。
両親に対しては、CEDの徴候に関する注意喚起を行い、多飲、黄疸といった疑わしい症状が現れた場合は、患児を担当する医療機関に連絡をとるようアドバイスしておく必要がある。
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「Clinical Trials.gov」、ならびにヨーロッパの「EU Clinical Trials Register」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
頭蓋外胚葉異形成症(CED)は常染色体潜性の遺伝形式をとる。
家族構成員のリスク
発端者の両親
- 罹患児の両親は絶対ヘテロ接合者である(すなわち、家族歴に基づいて、CEDを引き起こす病的バリアントを1つ有する保因者と目される)。
- 発端者の両親については、分子遺伝学的検査を行って、両者ともCEDを引き起こす病的バリアントをヘテロで有していることを確認し、信頼性のある再発リスク評価につなげることが推奨される。病的バリアントが片親からしか検出されない場合は、可能性として次のようなことを考慮する必要がある。
- 発端者で同定された病的バリアントのうちの1つが、発端者にde novoのイベントとして生じた可能性、もしくは、モザイクの片親において、接合後のde novoのイベントとして生じた可能性[Walczak-Sztulpaら2010,Jónssonら2017]。
- 病的バリアントを有しているほうの片親の染色体に片親性ダイソミーが生じて、発端者が病的バリアントのホモ接合を有するに至った可能性。
- ヘテロ接合者(保因者)は無症状で、本疾患を発症するリスクは有しない。
発端者の同胞
- 両親とも、CEDを引き起こす病的バリアントをヘテロで有していることが判明している場合、罹患者の同胞は、受胎の段階で、罹患者である可能性が25%、無症状の保因者である可能性が50%、罹患者でも保因者でもない可能性が25%である。
- CEDの臨床症候にはきわめて大きな幅がみられ、罹患同胞間で大きく異なる可能性がある。
- ヘテロ接合者(保因者)は無症状で、本疾患を発症するリスクは有しない。
発端者の子
CEDは稀な疾患で、長期的データも不足している。そのため、CED罹患者が生殖能力を有するかどうかという点については、今のところよくわかっていない。これまでCED罹患者が子をもうけたという報告はみられない。これは、CEDが重症度の高い疾患であるということ、ならびに、罹患者の寿命が短いことに起因するものと思われる。
他の家族構成員
両親ともCEDを引き起こす病的バリアントをヘテロで有していることが判明している場合、発端者の両親の同胞は、保因者であることに関し50%のリスクを有することになる。
保因者の検出
リスクを有する血族に対して保因者の検査を行うためには、家系内に存在するCEDを引き起こす病的バリアントを事前に同定しておくことが必要となる。
遺伝カウンセリングに関連した問題
早期診断・早期治療を目的としてリスクを有する血族に対して行う評価関連の情報については、「臨床的マネジメント」の「リスクを有する血縁者の評価」の項を参照されたい。
家族計画
- 遺伝的リスクの確定、出生前/着床前遺伝学的検査を受けるかどうかの話し合いに最も適しているのは、妊娠前の時期である。
- 罹患者、保因者、あるいは、保因者であるリスクを有する若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法であるとか、遺伝子・病原メカニズム・疾患等に対するわれわれの理解が、将来はより進歩していくことが予想される。そのため、分子診断の確定していない(すなわち、原因となった病原メカニズムが未解明の)発端者のDNAについては、保存しておくことを検討すべきである。より詳細な情報については、Huangら[2022]を参照されたい。
出生前診断ならびに着床前遺伝学的検査
分子遺伝学的検査
家系内に存在するCEDを引き起こす病的バリアントが同定済の場合は、出生前診断や着床前遺伝学的検査を行うことが可能である。
胎児超音波検査
第2三半期の超音波検査で、腎嚢胞、四肢の短縮、多指趾といったものが検出できることがある。
出生前検査の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。現在、多くの医療機関では、出生前検査を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- National Foundation for Ectodermal Dysplasias (NFED)
Phone: 618-566-2020
Email: info@nfed.org
www.nfed.org
- Ectodermal Dysplasias International Registry
Email: info@nfed.org
Ectodermal Dysplasias International Registry
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:頭蓋外胚葉異形成症:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specificデータベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| IFT43 | 14q24.3 | 繊毛内輸送タンパク質43ホモログ | IFT43 | IFT43 | |
| IFT52 | 20q13.12 | 繊毛内輸送タンパク質52ホモログ | IFT52 | IFT52 | |
| IFT122 | 3q21.3-q22.1 | 繊毛内輸送タンパク質122ホモログ | IFT122database | IFT122 | IFT122 |
| IFT140 | 繊毛内輸送タンパク質140ホモログ | IFT140@LOVD | IFT140 | IFT140 | |
| WDR19 | 4p14 | WDリピート包含タンパク質19 | WDR19@LOVD | WDR19 | WDR19 |
| WDR35 | 2p24.1 | WDリピート包含タンパク質35 | WDR35 | WDR35 |
データは、以下の標準資料から作成したものである。遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:頭蓋外胚葉異形成症関連のOMIMエントリー(閲覧はすべてOMIMへ)
| 218330 | CRANIOECTODERMAL DYSPLASIA 1; CED1 |
| 606045 | INTRAFLAGELLAR TRANSPORT 122; IFT122 |
| 608151 | WD REPEAT-CONTAINING PROTEIN 19; WDR19 |
| 613602 | WD REPEAT-CONTAINING PROTEIN 35; WDR35 |
| 613610 | CRANIOECTODERMAL DYSPLASIA 2; CED2 |
| 614068 | INTRAFLAGELLAR TRANSPORT 43; IFT43 |
| 614099 | CRANIOECTODERMAL DYSPLASIA 3; CED3 |
| 614378 | CRANIOECTODERMAL DYSPLASIA 4; CED4 |
| 614620 | INTRAFLAGELLAR TRANSPORT 140; IFT140 |
| 617094 | INTRAFLAGELLAR TRANSPORT 52; IFT52 |
分子レベルの病原
頭蓋外胚葉異形成症(CED)は、繊毛病として知られる疾患群のスペクトラムに属する疾患である[Baker & Beales 2009,Konstantinidouら2009,Arts & Knoers 2013]。繊毛病は、ほぼ人体のいたるところの細胞から突き出る繊毛(図2参照)の不具合に起因して生じる疾患と考えられている。微小管を中心に形成されるこの小器官は、ヒトの正常な発生、ならびに組織のホメオスタシスに不可欠な経路を調節するシグナル伝達ハブとして機能しているものと考えられている[Hildebarandtら2011]。
繊毛発生、ならびにシグナル伝達経路の調節には、繊毛輸送(繊毛内輸送[intraflagellar transport;IFT]とも呼ばれる)というプロセスが必要である。物質あるいはシグナル伝達分子の上向き(順行性)輸送は、ヘテロ三量体のキネシン2モーターの関与のもと、マルチサブユニットのIFT-B複合体が担い、下向き(逆行性)輸送は、ダイニン2モーターとIFT-A複合体が制御する[Hildebarandtら2011,Taschnerら2012]。
CED罹患者で現在までに同定されている病的バリアントの大多数は、六量体タンパク質であるIFT-A複合体の構成要素をコードする遺伝子内(IFT122〈以前の名称はWDR10〉,WDR35〈IFT121〉,WDR19〈IFT144〉,IFT43〈以前の名称はC14orf179〉,IFT140)に生じたものである[Gilissenら2010,Artsら2011,Bredrupら2011,Bacinoら2012,Hofferら2013,Linら2013,Caparrós-Martínら2015]。これら以外のIFT-A複合体構成メンバーをコードする遺伝子としてはTTC21Bがある。注目すべきは、IFT139、IFT140、DYNC2H1(これはダイニン2モーターのサブユニット)などのタンパク質をコードする遺伝子の変異によって引き起こされる疾患が、CEDと臨床症候が重なることである(「鑑別診断」の項を参照)[Dagoneauら2009,Davisら2011,Perraultら2012]。
CEDにおいてIFT-Aタンパク質複合体に不具合が生じると、線維芽細胞の繊毛の先端が膨らみ、正常であれば基底部から先端への(順行性)繊毛輸送を調節するIFT-B複合体タンパク質がそこに蓄積されることになる[Artsら2011,Bredrupら2011]。
IFT-Bタンパク質複合体の一部をコードするIFT52に生じた病的バリアントでもCEDが生じることが報告されている[Girishaら2016]。
CED罹患者の線維芽細胞においては、繊毛の短小化がみられるとの報告が存在する[Walczak-Sztulpaら2010]一方、必ずしもそうとは限らないとする報告もみられる[Bredrupら2011]。IFTに生じた不具合により繊毛の構造に異常が生じ、発生上重要なシグナル伝達カスケード(例えば、ヘッジホッグシグナル伝達経路)に障害が生じてCEDに至るものと考えられている[Walczak-Sztulpaら2010,Ocbinaら2011,Qinら2011,Liemら2012]。
疾患の発症メカニズム
機能喪失型である。
遺伝子特異的な検査技術上の考慮事項
遺伝子内縦列重複をもたらすIFT140の病的バリアントが同定されている。
更新履歴:
-
Gene Reviews著者: Philip J Ostrowski, MD and Katrina Tatton-Brown, MD.
日本語訳者: 佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2022.12.15. 日本語訳最終更新日: 22023.3.27.[in present]
![]()