常染色体顕性Robinow症候群
(AutosomalDominantRobinowSyndrome)
[Synonyms:FetalFaceSyndrome]
Gene Reviews著者:MaianRoifman,MD,HanBrunner,MD,JamieLohr,MD,JulianaMazzeu,PhD,andDavidChitayat,MD.
日本語訳者: 佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2019.10.3. 日本語訳最終更新日: 2023.4.18.
原文: AutosomalDominantRobinowSyndrome
要約
疾患の特徴
常染色体顕性Robinow症候群(ADRS)は、骨格所見(低身長,上肢を中心とした中間肢節短縮,短指趾)、性器奇形(男性では小陰茎/翼状陰茎,陰嚢低形成,停留精巣;女性では陰核・大陰唇の低形成)、顔面の形態異常(眼間開離/目立つ眼,前額部の突出,上向きの鼻孔,中顔面の後退)、歯の異常(咬合異常,叢生,部分性無歯症,永久歯萌出遅延)、二裂舌に加え、出生後も残る出生前大頭症が時としてみられることを特徴とする疾患である。これらより出現頻度の低い所見としては、腎奇形、橈骨頭脱臼、半椎や脊柱側彎をはじめとする脊椎奇形、爪の異形成、心奇形、口唇裂/口蓋裂、高次脳機能発達遅延(これは稀)などがある。心奇形が存在する場合は、これが罹病や死亡の主要原因となる。
Robinow症候群の1バリアントで、骨硬化症を伴うタイプのものは、DVL1の病的バリアントに起因して生じ、正常な身長、遺残性の大頭症、頭蓋骨の骨硬化を伴う骨密度上昇、難聴といったものが前述の典型的症候に加わることが特徴である。
診断・検査
発端者における常染色体顕性Robinow症候群の診断は、これを示唆する典型的所見がみられること、ないし、分子遺伝学的検査でDVL1、DVL3、WNT5Aのいずれかにヘテロ接合性の病的バリアントが同定されることをもって確定する。
臨床的マネジメント
症状に対する治療:
停留精巣、陰茎付着位置異常/陰茎前位陰嚢、口唇裂/口蓋裂に関しては、必要に応じ修正手術を行う。小陰茎の男性にはホルモン療法が有効な場合がある。通常、矯正歯科治療が必要となる。
定期的追跡評価:
乳児期から小児期にかけて、頭囲の計測を行う。発達評価を乳児期は3ヵ月ごと、その後は6-12ヵ月ごと、高次脳機能発達遅延がみられる場合は、必要に応じ、より高頻度に行う。歯科的評価を6-12ヵ月ごと、あるいは推奨に従った頻度で行う。小児期には定期的に聴覚評価を行う。異常が認められた場合は、必要に応じ通常の心臓や腎臓の評価を、それぞれの専門医の手で行う。
リスクを有する血縁者の評価:
治療や監視を開始することで利益が得られる可能性がある人を可能な限り早期に特定することを目的として、発端者の同胞について評価を行う。
妊娠に関する管理:
罹患女性は、妊娠に際して特段の合併症は生じないものと考えられる。罹患胎児については、異常胎位や児頭骨盤不均衡のため、帝王切開が必要になることがある。
遺伝カウンセリング
ADRSは、常染色体顕性の遺伝形式をとる。発端者には、病的バリアントが継承された例と、denovoの形で生じた例の両方がみられる。発端者の子は、病的バリアントを継承することに関し、50%の可能性を有する。ただ、分子遺伝学的検査の結果から、現れる臨床症候の重症度を予測することまではできない。家系内に存在するDVL1、DVL3、WNT5Aの病的バリアントが同定済の場合は、高リスクの妊娠に備えた出生前検査を行うことができる。
診断
本疾患を示唆する所見
以下に示すような臨床所見、家族歴を有する例については、常染色体顕性Robinow症候群(ADRS)を疑う必要がある[Mazzeuら2007,Personら2010,Beiraghiら2011,Roifmanら2015]。
本疾患を示唆する所見
臨床所見
骨格
- 低身長
- 上肢を中心とした中間肢節短縮
- 短指趾
生殖器
- 男性:小陰茎/翼状陰茎,陰嚢低形成,停留精巣
- 女性:陰核・大陰唇の低形成
頭蓋顔面
- 胎児の顔に似た顔面の変形:眼間開離と目立つ眼,前頭部毛髪線高位、前額部の突出,低い鼻梁,上向きの鼻孔を伴う短い鼻,広い鼻尖を伴う広い鼻梁,長い人中,中顔面の後退,耳介低位(図1ならびに図2参照)
- 咬合異常,歯の叢生と部分性無歯症,永久歯の萌出遅延,広い臼後部歯槽頂縁,歯槽頂縁の変形,分葉舌
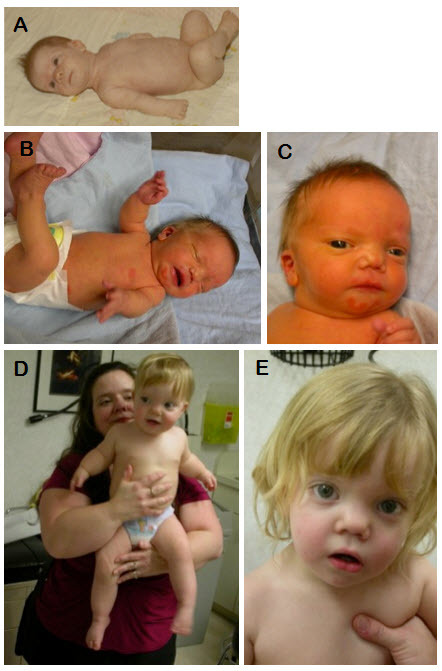
図1:ともにWNT5A関連常染色体顕性Robinow症候群である母と息子
A.罹患者である母の乳児期
B,C.罹患男児の出生時
D.母親(39歳)と息子(2歳)
E.息子の3歳時
眼間開離と目立つ眼、前頭部毛髪線高位、前額部の突出、低い鼻梁、上向きの鼻孔を伴う短い鼻、広い鼻尖を伴う広い鼻梁、長い人中、中顔面の後退、耳介低位、上肢を中心とした四肢短縮に注目されたい。
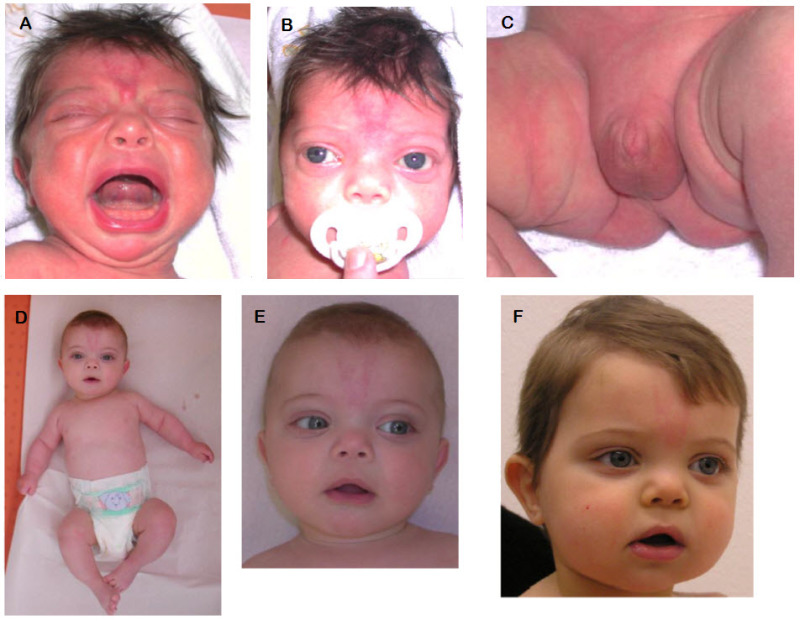
図2:WNT5A関連常染色体顕性Robinow症候群の1男児の異なる年齢の写真
眼間開離と目立つ眼,前頭部毛髪線高位,前額部の突出,低い鼻梁,上向きの鼻孔を伴う短い鼻,広い鼻尖を伴う広い鼻梁,長い人中,耳介低位(A,B,C,D),性器低形成(C),上肢を中心とした四肢の短縮(D)に注目されたい。
家族歴
常染色体顕性遺伝に一致した家族歴を示す。
注:常染色体顕性遺伝の家族歴が判然としない場合でも、本疾患の可能性が否定されるわけではない。
診断の確定
発端者における常染色体顕性Robinow症候群の診断は、これを示唆する特徴的な所見がみられること、ないし、分子遺伝学的検査でDVL1、DVL3、WNT5Aのいずれかにヘテロ接合性の病的バリアント(pathogenicとlikelypathogenicの両方を含む)が同定されることをもって確定する(表1参照)。
注:(1)DVL1、DVL3、WNT5Aのいずれにもヘテロ接合性の病的バリアントがみられない場合は、ROR2もしくはNXNの両アレル性の病的バリアント(常染色体潜性Robinow症候群の原因)、あるいは、FZD2のヘテロ接合性の病的バリアント(常染色体顕性肩骨異形成症2型の原因)が存在する可能性を排除しておくことが望ましい。
(2)アメリカ臨床遺伝ゲノム学会(ACMG)のバリアントの解釈に関するガイドラインによると、「pathogenic」のバリアントと「likelypathogenic」のバリアントとは臨床の場では同義であり、ともに診断に供しうるものであると同時に、臨床的な意思決定に使用しうるものとされている。本セクションで「病的バリアント」と言うとき、それは、あらゆるlikelypathogenicのバリアントまでを包含するものと理解されたい。
分子遺伝学的検査のアプローチとしては、遺伝子標的型検査(同時施行あるいは直列型で行う単一遺伝子検査、マルチ遺伝子パネル)と網羅的ゲノム検査(エクソームシーケンシング,ゲノムシーケンシング)を組み合わせるやり方が考えられる。
- 単一遺伝子検査
配列解析では、遺伝子内の小欠失/挿入や、ミスセンス・ナンセンス・スプライス部位バリアントなどが検出されるが、エクソン単位あるいは遺伝子全体の欠失/重複といったものは検出されない。
DLV1とDLV3の配列解析を最初に行う。WNT5Aの配列解析は、これらと同時に行うか、もしくは、DLV1やDLV3に病的バリアントが検出されないことを受けて行うようにする。
注:DLV1とDLV3について報告されている疾患関連バリアントはすべて、最後尾とその手前のエクソン(エクソン14と15)に生じたフレームシフトバリアントである。
- マルチ遺伝子パネル
現況の表現型と直接関係のない遺伝子の意義不明バリアントや病的バリアントの検出を抑えつつ、疾患の遺伝的原因の特定に最もつながりやすいのは、DLV1、DLV3、WNT5A、FZD2、ROR2、NXNその他の関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネルであるように思われる。
注:(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。DVL1とDVL3については、エクソン14と15がアッセイで十分にカバーされていることを確認しておくことが必要である。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝学的検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
- 網羅的ゲノム検査
利用可能なようであれば、エクソームシーケンシングやゲノムシーケンシングなどの、より包括的なゲノム検査も検討対象になりうる。そうした検査を行うことで、それまで検討されていなかった病名(例えば、検討対象になっていたものとは別の遺伝子の変異により、臨床症候の点ではADRSと似た状態が引き起こされるといった状態)が判明あるいは示唆される場合がある。
表1:常染色体顕性Robinow症候群で用いられる分子遺伝学的検査
| 遺伝子1,2 | その遺伝子の病的バリアントが常染色体顕性Robinow症候群の中で占める割合 | 配列解析で病的バリアント3が検出される割合4,5 |
|---|---|---|
| DVL1 | 発端者16人6(検査した発端者総数は明らかでない) | 99%超7 |
| DVL3 | 発端者7人8(検査した発端者総数は明らかでない) | 99%超7 |
| WNT5A | 発端者8人9(検査した発端者総数は明らかでない) | 99%超 |
| 不明10,11 | 利用不可 |
- 遺伝子の記載はアルファベット順。
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- この遺伝子で同定されているアレルバリアントの情報については、「分子遺伝学」の項を参照のこと。
- 配列解析を行うことで、benign、likelybenign、意義不明、likelypathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、遺伝子内の小欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。
- 配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
- ADRSは機能獲得型のメカニズムで発症すること、ならびに、これまでに遺伝子内の大きな欠失や重複が報告されていないことから、遺伝子内の欠失/重複解析によって疾患の原因となるようなバリアントが検出される可能性は低い。
- Bunnら[2015],Whiteら[2015],Whiteら[2016],Whiteら[2018]
- 現段階で同定されている病的バリアントはすべてフレームシフトバリアントである。
- Whiteら[2016],Danyelら[2018],Whiteら[2018]
- Personら[2010],Roifmanら[2015],Xiongら[2016],Whiteら[2018]
- Robinow症候群の臨床診断を受けた孤発例で、RAC3ならびにGPC4の病的バリアントが報告された例が存在する[Whiteら2018]。
- FZD2の病的バリアントにより生じる肩骨異形成症2型は、ADRSと多くの臨床症候を共有する。この2つの疾患が同一の表現型スペクトラムの一部分であるか否かという点については、明らかではない(「鑑別診断」の項を参照)。
臨床的特徴
臨床像
常染色体顕性Robinow症候群(ADRS)は、通常、低身長、上肢を中心とした中間肢節短縮、短指趾が現れる骨系統疾患である。本疾患を疑わせる奇形は、その他にもさまざまな種類(現れ方も多様)のものがある。
顔面
ADRSでみられる頭蓋顔面症候については、「本疾患を示唆する所見」の項に要約を述べた通りである。これらの症候は、出生時あるいは幼児期に最もはっきり認められる。特徴的顔面症候は、年齢とともに目立たなくなっていく。
- 大頭症以外に、成人期にもみられる顕著な顔面症候としては、眼間開離、広い鼻梁、広い鼻尖などがある。
- 咬合異常は、幼児期には明らかに認められるようになり、永久歯列にも影響が及び、成人期まで存続する。
乳歯列が18歳まで続き、抜歯を要した1例が報告されている[Roifmanら2015]。
骨格
低身長は、出生時にはほぼ全例で認められるが、時には胎生20週の段階で超音波検査で同定されるような例もみられる[Mazzeuら2007,Castroら2014,Roifmanら2015]。
- 低身長は成人期にまで存続するものの、通常は重度ではなく、成人期の最終身長は、大多数の例では-2SDか、それをわずかに下回る程度である[Mazzeuら2007,Personら2010,Roifmanら2015]。
- DVL1関連ADRS罹患者の中には、骨格に独特の表現型が現れる例がみられる(「遺伝子ごとにみた表現型との相関」の項を参照)。
泌尿生殖器
男女とも、性器低形成が出生の段階で明らかに認められる。
- 小陰茎がみられる場合があるものの、中には、陰茎の大きさそのものは正常で、翼状陰茎あるいは陰嚢組織中への埋没のため、あるいは、陰茎の脚部の位置が下後方にずれて坐骨結節内側面から出ているために小さく見えるような例もみられる[Wilcoxら1997]。こうしたものについては美容的再建術の対象となろう(「臨床的マネジメント」の項を参照)。
- 小陰茎は、ADRSにおいては、多くみられるといった程度の頻度の症候であるように思われる(なお、常染色体潜性Robinow症候群では必発症候である)。
- ADRSにおける陰茎前位陰嚢の発生頻度は、現時点では明らかではない。
思春期ならびに妊孕性
著者らの知る限り、思春期や妊孕性に異常はみられず、妊娠した罹患女性は満期まで妊娠を継続できる。ただ、分娩にあたっては、児頭骨盤不均衡のため帝王切開を要する場合がある。
心異常
ADRS罹患者の少数(25%未満)に心異常がみられる。
- Robinow症候群(顕性型と潜性型の両方を含む)で報告されている心奇形としては、肺動脈弁狭窄/閉鎖、心房中隔欠損、心室中隔欠損、大動脈縮窄、Fallot四徴、三尖弁閉鎖などがある[Webberら1990,Al-Ataら1998]。
- 心奇形が存在する場合は、これが罹病や死亡の主要原因となる。
難聴(両側性混合性)
DVL1関連ADRS罹患者の一部で、両側性混合性難聴が報告されている[Bunnら2015,Whiteら2015]。
臍ヘルニア
DVL1関連ADRS罹患者の一部で、臍ヘルニアが報告されている[Whiteら2015]。
知能
知能はふつう正常であるが、ADRS罹患者の少数に高次脳機能発達遅延がみられる。
比較的出現頻度の低いその他の症候(25%未満)[Mazzeuら2007,Personら2010,Roifmanら2015]
- 腎奇形(通常は水腎症)
- 橈骨頭脱臼
- 脊椎奇形と脊柱側彎
- 抜去を要する乳歯の残存
- 爪の異形成
- 口唇裂/口蓋裂
遺伝子ごとにみた表現型との相関
DVL1
DVL1関連ADRS罹患者の中のある1つのサブセットは、正常低値の最終身長、頭蓋骨硬化を伴う骨密度上昇、大頭症(+2.5SDから+6SD超の範囲)を示す[Bunnら2015,Whiteら2015]。
DVL3
DVL3関連ADRS罹患者4人中3人に心異常が確認されている。
骨硬化の報告はみられない。
WNT5A
WNT5A関連ADRSの5家系で、WNT5Aタンパク質のドメイン特異的病的バリアントが同定されており、これがADRSの典型症候(特徴的顔貌,低身長,中間肢節短縮,性器低形成)を伴う臨床的表現型を引き起こす変異であると考えられている[Roifmanら2015]。
遺伝型-表現型相関
はっきりした遺伝型-表現型相関は知られていない。
発生頻度
ADRSは非常に稀である。本疾患の正確な発生頻度はわかっていない。文献で報告されたADRSの数は、80家系に満たない。
遺伝子学的に関連のある疾患(同一アレル疾患)
DLV1、DLV3、WNT5Aの病的バリアントに起因するものとしては、本GeneReviewで述べたもの以外の表現型は知られていない。
鑑別診断
ROR2関連Robinow症候群
ROR2関連Robinow症候群は、ROR2の両アレル性病的バリアントに起因する常染色体潜性遺伝の骨系統疾患である。ADRSに類似した症候としては、特徴的顔貌、低身長、中間肢節短縮、性器低形成などがある。ROR2関連Robinow症候群でみられる症候は、ADRSより概して重度で、腎奇形、先天性心疾患、脊椎奇形、肋骨癒合、脊柱側彎、高次脳機能発達遅延といったものがADRSより高頻度に現れる。ROR2関連Robinow症候群の鑑別上の特徴は、主として拇指に現れる末節骨の裂である。
NXN関連Robinow症候群(OMIM618529)
こちらも常染色体潜性の継承を示し、互いに血縁関係のない2家系3人でNXNの両アレル性病的バリアントが報告されている。3人とも、典型的頭蓋顔面症候、中間肢節短縮、短指趾をはじめとするRobinow症候群の古典的臨床所見を有していた[Whiteら2018]。1人は、血族結婚の両親から生まれた例で、NXNのナンセンスバリアントのホモ接合であった。もう1つの家系の2同胞は、NXNの病的バリアントを複合ヘテロで有していた。
注:NXNタンパク質は、Robinow症候群の発生に密接に係わるWNT5Aシグナル伝達経路における結合パートナータンパク質である。ROR2はWNT5Aと結合し、FZD2と相互作用を行う。この相互作用の結果はdisheveledタンパク質(DVL1,DVL3)へと送られ、NXNによってさらなる安定化を受ける。この複合体は、細胞骨格の再構成や細胞極性に関与するJNKシグナル伝達を活性化する。
Aarscog症候群(OMIM305400)
Aarscog症候群は、FGD1の変異によって引き起こされるX連鎖性疾患である。その顔面症候(高い前頭部毛髪線,前額部の突出,眼間開離,上向きの鼻孔)は、ADRSと類似している。肺動脈弁狭窄の報告もみられる。ADRSでみられる性器低形成が広いスペクトラムを示すのに対し、Aarscog症候群男性でみられる性器低形成は襟巻状陰嚢が特徴である。その他の特徴的症候としては、widow’speak(訳注:一般的には「富士額」と訳されるが、単なる生え際のラインが作る曲線の形ではなく、跳ね上がったような毛髪の向きの乱れを伴うものを指す)と靱帯弛緩が挙げられる。
ADRSでみられる脊椎奇形や歯の萌出遅延は、Aarscog症候群ではみられない。Aarscog症候群でみられる特徴的な四肢奇形としては、短指趾、合指趾、第5指の彎指などがある。
常染色体顕性OpitzG/BBB症候群(ADOS)とX連鎖性OpitzG/BBB症候群(XLOS)
常染色体顕性OpitzG/BBB症候群(ADOS)とX連鎖性OpitzG/BBB症候群(XLOS)は、それぞれ、22q11.2の欠失、MID1の変異によって生じる疾患である。ADRSと類似するものとしては、顔面症候(高い前頭部毛髪線,前額部の突出,眼間開離,広い鼻梁,上向きの鼻孔)、ならびに尿性器奇形(尿道下裂,停留精巣,陰嚢低形成/二分陰嚢)がある。XLOSにおいては、同時に、喉頭気管食道奇形(これはADRSではみられない)、脳奇形、発達遅滞、口唇裂/口蓋裂(これはADOSやXLOS[罹患者の50%]のほうがADRSよりはるかに多くみられる)がみられることも特徴である。ADOSやXLOSでは通常、低身長や中間肢節短縮はみられない。
軟骨無形成症
軟骨無形成症は、FGFR3の変異に起因して生じる常染色体顕性遺伝疾患である。軟骨無形成症に特徴的に現れる顔面症候は、ADRSのそれ(大頭症,高い前頭部毛髪線と前額部の突出,低い鼻梁,尖った鼻,中顔面の後退)と類似している。軟骨無形成症罹患者は、ADRSより頭囲が大きく、大頭症は生涯を通じて続く。また、水頭症の発生率も高い。軟骨無形成症に特徴的な骨格症候としては、三尖手、乳児期における腰椎の突背と筋緊張低下、脊椎前彎、小児期後期における下肢の彎曲、すべての長管骨に現れるより重度の短縮などがある。眼間開離は軟骨無形成症においてはみられない。
肩骨異形成症2型(OMIM164745)
肩骨異形成症2型は、特徴的な骨格所見、男性器の低形成(小陰茎,尿道下裂,停留精巣など)、顔面の形態異常を呈する稀な常染色体顕性遺伝疾患である。ADRSと明確に異なる症候としては、正常な身長、上肢近位肢節の短縮、第1中手骨の短縮、関節頭の低形成を伴う上腕骨の短縮などがある。下肢に異常はみられない。顔面症候としては、前額部の突出、二分鼻尖を伴う低い鼻梁、長い人中などがある。肩骨異形成症2型罹患者に眼間開離はみられない。肩骨異形成症2型の4家系で、FZD2の病的バリアントのヘテロ接合が同定されている[Whiteら2018]。肩骨異形成症2型とRobinow症候群との間に表現型の重なりがみられることから、肩骨異形成症2型というのは、実際にはRobinow症候群の臨床的スペクトラム内に属するものであって、短い上腕骨と橈骨頭脱臼が出現しやすい1つのサブタイプであろうとの仮説を唱える向きもある。
臨床的マネジメント
最初の診断に続いて行う評価
常染色体顕性Robinow症候群(ADRS)と診断された罹患者については、疾患の範囲やニーズを把握するため、診断に至る過程ですでに実施済ということでなければ、表2にまとめたような評価を行うことが推奨される。
表4:常染色体顕性Robinow症候群罹患者の最初の診断後に行うことが推奨される評価
| 系/懸念事項 | 評価 | コメント |
|---|---|---|
| 頭蓋顔面 | 口腔顔面裂の存在に関する臨床的評価 | 頭蓋顔面チームへの紹介を検討する。 |
| 矯正歯科の相談 | 歯の不整あるいは乳歯の残存に関して。 | |
| 耳 | 聴覚評価 | |
| 心血管 | 心エコー | 先天性心疾患の評価目的。 |
| 泌尿生殖器 | 腎超音波 | 腎奇形や水腎症の評価目的。 |
| 女性:骨盤超音波 | ミューラー管奇形の評価目的。 | |
| 男性:陰茎の位置/陰茎・陰嚢の位置関係の異常、ならびに停留精巣に関する評価 | 小陰茎がみられる場合は、ホルモン治療の可能性を考慮して内分泌内科医との相談を検討する。 | |
| 筋骨格 | 四肢、胸、脊椎、頭蓋のX線写真 | 骨格病変の範囲の確定目的。 |
| 神経 | 発達遅滞に関する評価 | 遅滞ありの場合は、正式な神経精神医学的検査や高次脳機能検査を目的とした紹介。 |
| その他 | 臨床遺伝医ないし遺伝カウンセラーとの面談 |
症候に対する治療
表3:常染色体顕性Robinow症候群罹患者の症候に対する治療
| 症候/懸念事項 | 治療 | 考慮事項/その他 |
|---|---|---|
| 口唇裂/口蓋裂 | 外科的改善 | 多職種の頭蓋顔面チームによる管理が推奨される。 |
| 歯列不整あるいは乳歯の残存 | 矯正歯科の標準治療 | |
| 難聴 | 標準治療 | 「遺伝性難聴・聴力喪失概説」のGeneReviewを参照 |
| 先天性心疾患 | 心臓病専門医ないし心胸郭外科医による標準治療 | |
| 停留精巣 | 精巣固定術 | |
| 陰茎の位置/陰茎・陰嚢の位置関係の異常 | 泌尿器科医へ紹介 | 外科的に改善可能かどうかに関する検討。 |
| 小陰茎 | ホルモン療法を検討1 | 内分泌内科医へ紹介。 |
- 重度の小陰茎を呈する3人の男児で、ヒト絨毛性ゴナドトロピンとテストステロンの注射により陰茎の長さや精巣の体積に改善がみられている[Solimanら1998]。
定期的追跡評価
表4:常染色体顕性Robinow症候群罹患者で推奨される定期的追跡評価
| 系/懸念事項 | 評価 | 実施頻度 |
|---|---|---|
| 頭蓋顔面 | 歯科的評価 | 歯科医の手で6ヵ月から1年ごと。 |
| 耳 | 聴覚評価 | 小児期に。 |
| 心血管 | 心病変を伴う例について標準的モニタリング | 心臓病専門医の手で行う。 |
| 泌尿生殖器 | 腎病変を伴う例について標準的モニタリング | 泌尿器科医の手で行う。 |
| 神経 | 発達の進行状況の評価 | 小児期/思春期には来院ごと。 |
リスクを有する血縁者の評価
発端者の同胞については、治療や監視を行うことで利益が得られる人を可能な限り早期に特定することを目的とした評価を行うことが望ましい。家系内に存在するDVL1、DVL3、WNT5Aの病的バリアントが既知の場合は、リスクを有する同胞の遺伝学的状態を明確にするための分子遺伝学的検査を行うことができる。
リスクを有する血縁者に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
妊娠に関する管理
罹患女性については、妊娠に際して特段の合併症は生じないものと思われる。罹患胎児については、異常胎位や児頭骨盤不均衡のため、帝王切開が必要になることがある。帝王切開を要した骨盤位のADRSの1例が報告されている[Roifmanら2015]。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「ClinicalTrials.gov」、ならびにヨーロッパの「EUClinicalTrialsRegister」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
DVL1、DVL3、WNT5Aの病的バリアントに起因して生じるRobinow症候群は、常染色体顕性の遺伝形式をとる。
家族構成員のリスク
発端者の両親
- 常染色体顕性Robinow症候群(ADRS)と診断された罹患者の一部は、罹患者である親からの継承例である。
- ADRS発端者の中には、denovoの病的バリアントに起因して本疾患に至った例もみられる。denovoの病的バリアントに起因する例の比率は、明らかになっていない。
- 見かけ上denovoと思われるDVL1、DVL3、WNT5Aの病的バリアントをもつ発端者の両親については、関連する臨床症候の有無を調べる身体の詳細な診査、ならびに発端者で同定された病的バリアントを調べる検査を行うことが推奨される。
- 発端者で同定されたDVL1、DVL3、WNT5Aの病的バリアントが両親いずれの白血球DNAからも検出されない、ないし、どちらの親も本疾患を窺わせる臨床症候を有しないという場合、その病的バリアントはdenovoのものである可能性が高い。別の可能性として、生殖細胞系列モザイクの片親からの継承により発端者が疾患を有するに至ったということも考えられる。
- 両親に対して詳しい評価を行ってみると、実際は片親が罹患者であるにもかかわらず、表現型が軽度であったためにそれまで診断をすり抜けていたことが明らかになるようなことがある。したがって、見かけ上、家族歴陰性であるように思われても、適切な評価を行うまでは、家族歴陰性の確定はできない。
- 注:最初に病的バリアントが生じたのが片親のほうだったということであれば、片親はそのバリアントを体細胞モザイクで有していたために、症候が軽度ないしごく軽微なものにとどまったことが考えられる[Whiteら2018]。
発端者の同胞
発端者の同胞の有するリスクは、発端者の両親の遺伝的状態によって変わってくる。
- 発端者の片親が罹患者であった場合、同胞の有するリスクは50%である。ただ、その場合に生じる臨床症候の重症度については、分子遺伝学的検査の結果から予測することはできない。
- 両親とも臨床的には非罹患者と目される場合であれば、発端者の同胞の有するリスクは低いものと思われる。
- 発端者で同定されたDVL1、DVL3、WNT5Aの病的バリアントが両親いずれの白血球DNAからも検出されない場合、同胞の有するリスクは一般集団より若干高い(それでも1%未満)程度と推定される。それは、理論上、親の生殖細胞系列モザイクの可能性が残るからである。
発端者の子
ADRS罹患者の子は、50%の確率で病的バリアントを継承することになる。ただ、その場合に生じる臨床症候の重症度については、分子遺伝学的検査の結果から予測することはできない。
他の家族構成員
他の血縁者の有するリスクは、発端者の両親の状態によって変わってくる。仮に、片親が罹患者であったということになれば、その片親の血族にあたる人はすべてリスクを有することになる。
関連する遺伝カウンセリング上の諸事項
早期診断・早期治療を目的としてリスクを有する血族に対して行う評価関連の情報については、「臨床的マネジメント」の「リスクを有する血縁者の評価」の項を参照されたい。
見かけ上denovoと思われる病的バリアントを有する家系についての考慮事項
常染色体顕性遺伝疾患を有する発端者のいずれの親も病的バリアントを有していない、また、その疾患の臨床症候も有していないということであれば、発端者のもつその病的バリアントはdenovoのものである可能性が濃厚である。ただ、代理父、代理母(例えば生殖補助医療によるもの)、もしくは秘匿型の養子縁組といった医学とは別次元の理由が潜んでいる可能性についても、併せて調べてみる必要がある。
家族計画
- 遺伝的リスクの確定、出生前検査を受けるかどうかの話し合いといったことに最も適しているのは、妊娠前の時期である。
- 罹患者である若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法であるとか、遺伝子・病原メカニズム・疾患等に対するわれわれの理解が、将来はより進歩していくことが予想される。そのため、分子診断の確定していない(すなわち、原因となった病原メカニズムが未解明の)発端者のDNAについては、保存しておくことを検討すべきである。
出生前検査ならびに着床前遺伝学的検査
分子遺伝学的検査
家系内に存在するDVL1、DVL3、WNT5Aの病的バリアントが同定済の場合は、高リスクの妊娠に備えた出生前検査や着床前遺伝学的検査を行うことが可能である。ただ、生じる臨床症候の重症度については、分子遺伝学的検査の結果から予測することはできない。
胎児超音波検査
妊娠20週の段階での胎児超音波検査で、長管骨の短縮(長さが-2SD)、大頭症、特徴的顔面症候(眼間開離,広い鼻梁,平坦な前頭骨)といったものが検出される場合がある[Castroら2014,Roifmanら2015]。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- HumanGrowthFoundation
- LittlePeopleofAmerica
- MAGICFoundation
- RestrictedGrowthAssociation(RGA)
Phone:888-LPA-2001;714-368-3689
Fax:707-721-1896
Email:info@lpaonline.org
www.lpaonline.org
Phone:800-362-4423;630-836-8200
Fax:630-836-8181
Email:contactus@magicfoundation.org
www.magicfoundation.org
POBox15755
SolihullB933FY
UnitedKingdom
Phone:+4403001111970
Fax:+4403001112454
Email:office@restrictedgrowth.co.uk
www.restrictedgrowth.co.uk
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:常染色体顕性Robinow症候群:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | HGMD | ClinVar |
|---|---|---|---|---|
| DVL1 | 1p36.33 | Segment polarity protein dishevelled homolog DVL-1 | DVL1 | DVL1 |
| DVL3 | Segment polarity protein dishevelled homolog DVL-3 | DVL3 | DVL3 | |
| WNT5A | 3p14.3 | Protein Wnt-5a | WNT5A | WNT5A |
データは、以下の標準資料から作成したものである。
遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。
リンクが張
られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:常染色体顕性Robinow症候群関連のOMIMエントリー(内容の閲覧はOMIMへ)
| 164975 | WINGLESS-TYPE MMTV INTEGRATION SITE FAMILY, MEMBER 5A; WNT5A |
| 180700 | ROBINOW SYNDROME, AUTOSOMAL DOMINANT 1; DRS1 |
| 601365 | DISHEVELLED 1; DVL1 |
| 601368 | DISHEVELLED 3; DVL3 |
| 616331 | ROBINOW SYNDROME, AUTOSOMAL DOMINANT 2; DRS2 |
| 616894 | ROBINOW SYNDROME, AUTOSOMAL DOMINANT 3; DRS3 |
分子レベルの病原
Wntファミリーは、胚のパターン形成、細胞の分化・増殖・移動といった形態形成上の重要なイベントを調節するタンパク質である。Wntシグナル伝達経路は、βカテニンの関与する古典的(canonic)経路と、βカテニンとは無関係に作用する非古典的(non-canonic)経路とに分けられる。
WNT5Aは、古典的経路と非古典的経路の両方において機能し、細胞移動を要する発生プロセスにおいて必須のWntファミリーメンバーである(Nishitaら[2010]によるレビューがある)。WNT5Aは、オーファンチロシンキナーゼ受容体であるROR2のコレセプターとして知られる[Oishiら2003,Mikels&Nusse2006,Schambony&Wedlich2007]。このことは、WNT5A関連Robinow症候群とROR2関連Robinow症候群との間に臨床的表現型の重なりがみられる背景を説明するものである。DVL1とDVL3は、segmentpolarityproteindishevelledホモログであるDVL-1とDVL-3をコードしている。この2つのタンパク質は、Wntシグナル伝達経路、特にWnt5a-ROR2非古典的経路において必須の下流メディエーターである[Whiteら2015,Whiteら2016]。
DVL1
遺伝子構造
DVL1は15のエクソンから成る。
病的バリアント
これまでにわかっているADRS関連のバリアントは、すべてエクソン14と15のフレームシフトバリアントである(表5に示した例を参照)[Bunnら2015,Whiteら2015,Whiteら2016]。メカニズムは機能獲得型であるとされており(「異常遺伝子産物」の項を参照)、これらのバリアントは最後のエクソンならびに最後から2番目のエクソンに位置していることから、ナンセンス変異依存mRNA分解機構を免れるトランケーション型の転写産物を産生する結果となっていることが示唆される。
表5:DVL1の代表的な病的バリアント
| DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | 参照配列 |
|---|---|---|
| c.1505_1517del13 | p.His502ProfsTer143 | NM_004421.2 NP_004412.2 |
| c.1508delC | p.Pro503ArgfsTer146 | |
| c.1519delT | p.Trp507GlyfsTer142 | |
| c.1529delG | p.Gly510ValfsTer139 | |
| c.1562delC | p.Pro521HisfsTer128 | |
| c.1570_1571delinsC | p.Phe524ProfsTer125 | |
| c.1576_1583delinsG | p.Pro526AlafsTer121 | |
| c.1615delA | p.Ser539AlafsTer110 |
表中のバリアントは、著者の提供したものをそのまま載せたもので、GeneReviewsのスタッフが独立した立場でバリアントの分類を確認したものではない。GeneReviewsは、HumanGenomeVariationSociety(varnomen.hgvs.org)の標準命名規則に準じた表記を行っている。命名法の説明に関しては、「QuickReference」を参照されたい。
正常遺伝子産物
DVL1はWnt受容体の下流に位置する細胞内足場タンパク質であるdishevelledファミリーのメンバーである。DVL1は、古典的経路、非古典的経路両方のWntシグナル伝達に一定の役割を果たしている(OMIM601365)。
異常遺伝子産物
エクソン14と15のフレームシフトバリアントによって生じる異常遺伝子産物は機能獲得型あるいはドミナントネガティブのメカニズムでWntシグナル伝達を障害すると考えられており、これが、正常な身長、大頭症、骨密度の上昇といった独特の骨格表現型の背景になっているものと考えられている[Whiteら2015]。
DVL3
遺伝子構造
DVL3は15のエクソンから成る。
病的バリアント
DVL1と同様、DVL3の場合もADRS関連バリアントの大多数はエクソン14と15のフレームシフトバリアントである(表6に示した例を参照)[Whiteら2015,Whiteら2016]。イントロン14のスプライス受容部位のバリアントも報告されている[Whiteら2016]。メカニズムは機能獲得型とされており(「異常遺伝子産物」の項を参照)、これらのバリアントは最後のエクソンならびに最後から2番目のエクソンに位置していることから、ナンセンス変異依存mRNA分解機構を免れるトランケーション型の転写産物を産生する結果となっていることが示唆される。
表6:DVL3の代表的な病的バリアント
| DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | 参照配列 |
|---|---|---|
| c.1585delG | p.Ala529ProfsTer139 | NM_004421.2 NP_004412.2 |
| c.1617delG | p.Gln539HisfsTer129 | |
| c.1715-2A>G | p.? | |
| c.1715-1G>A | p.? | |
| c.1716delC | p.Ser573ValfsTer95 | |
| c.1749delC | p.Ser583ArgfsTer85 |
表中のバリアントは、著者の提供したものをそのまま載せたもので、GeneReviewsのスタッフが独立した立場でバリアントの分類を確認したものではない。GeneReviewsは、HumanGenomeVariationSociety(varnomen.hgvs.org)の標準命名規則に準じた表記を行っている。命名法の説明に関しては、「QuickReference」を参照されたい。
正常遺伝子産物
DVL3はWnt受容体の下流に位置する細胞内足場タンパク質であるdishevelledファミリーのメンバーである。DVL3は、古典的経路、非古典的経路両方のWntシグナル伝達に一定の役割を果たしている。
異常遺伝子産物
エクソン14と15のフレームシフトバリアントによって生じる異常遺伝子産物は機能獲得型あるいはドミナントネガティブのメカニズムでWntシグナル伝達を障害すると考えられており、これが、正常な身長、大頭症、骨密度の上昇といった独特の骨格表現型の背景になっているものと考えられている[Whiteら2015]。
WNT5A
遺伝子構造
WNT5Aは5つのエクソンから成る。選択的スプライシングにより複数の転写バリアントが生じる。遺伝子ならびにタンパク質に関する情報の詳細は、表Aの「遺伝子」の欄を参照のこと。
病的バリアント
これまでに、常染色体顕性Robinow症候群の5家系で、複数のミスセンスバリアント(表7)、1つのインフレームの重複、1つのインフレームの欠失/重複が同定されている[Personら2010,Roifmanら2015,Whiteら2018]。
表7:WNT5Aの代表的な病的バリアント
| DNAヌクレオチドの変化 (別表記1) | 予測されるタンパク質の変化 (別表記1) |
参照配列 |
|---|---|---|
| c.206G>A | p.Cys69Tyr | NM_003392.4 NP_003383.2 |
| c.248G>C | p.Cys83Ser | |
| c.257A>G2 | p.Tyr86Cys | |
| c.544_545delinsTC (544-545CT>TC) |
p.Cys182Ser (Cys182Arg)* |
*訳注:上の行では「Ser」、下の行では「Arg」となっているが、どちらか一方もしくは両方が誤っているのではないかと思われる。それぞれ「p.Leu182Ser」、「(Leu182Ser)」の誤りである可能性が考えられる。
表中のバリアントは、著者の提供したものをそのまま載せたもので、GeneReviewsのスタッフが独立した立場でバリアントの分類を確認したものではない。GeneReviewsは、HumanGenomeVariationSociety(varnomen.hgvs.org)の標準命名規則に準じた表記を行っている。命名法の説明に関しては、「QuickReference」を参照されたい。
- 現在の命名規則とは異なるバリアントの表記
- 2家系がこのバリアントを有していた。
正常遺伝子産物
WNT5Aは、細胞移動を要する発生プロセスにおいて、決定的に重要である[Nishitaら2010によるレビューがある]。WNT5Aの三次元構造については、未だよくわかっていない。
四肢の伸長のコントロールに、肢芽中のWNT5Aシグナル伝達勾配が係わっていることが明らかになっている[Gaoら2011]。また、WNT5Aのマウスオーソログがマウスモデルにおける性腺発生のいくつかの段階を制御していることも明らかになっている[Tevosian2012]。
異常遺伝子産物
Roifmanら[2015]は、WNT8のフォールディングを実験的に解いたものを用いて行ったWNT5A相同モデルに基づいて、WNT5A関連ADRS罹患者でみられる病的バリアントはすべてタンパク質の片側のみに位置していると思われることを明らかにし、その上でこれらのバリアントが、正常な複合体形成ないしタンパク質内結合を障害する形で、Wnt経路における他のタンパク質との相互作用に影響を及ぼしている可能性が考えられるとしている。WNT5A関連疾患の病原メカニズムは未だ明らかではないものの、疾患関連のインフレームのミスセンスバリアントがタンパク質の特定の領域に固まって存在していることを示す一連の報告から、タンパク質のもつ機能の変化が疾患のメカニズムとして関与している可能性が考えられる。
更新履歴:
-
Gene Reviews著者:MaianRoifman,MD,HanBrunner,MD,JamieLohr,MD,JulianaMazzeu,PhD,andDavidChitayat,MD.
日本語訳者: 佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2019.10.3. 日本語訳最終更新日: 2023.4.18.[in present]
![]()