色素失調症
(Incontinentia Pigmenti)
[Synonyms:Bloch-Sulzberger Syndrome]
Gene Reviews著者: Angela E Scheuerle, MD, FAAP, FACMG and Matilde Valeria Ursini, PhD.
日本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(カレス記念病院ゲノム医療センター)
GeneReviews最終更新日: 2025.4.3. 日本語訳最終更新日: 2025.4.6.
要約
疾患の特徴
色素失調症(IP)は、皮膚、毛髪、歯、爪、眼、中枢神経系に影響を及ぼす疾患である。
基本的に女性が罹患するが、時に男性にも現れる。特徴的皮膚病変が、次の4段階で進んでいく。
Ⅰ.水疱期(出生から生後約4ヵ月まで)
Ⅱ.疣状発疹期(数ヵ月間)
Ⅲ.渦巻状色素沈着期(生後約6ヵ月から成人期まで)
Ⅳ.線状色素消退期
脱毛、無歯症、歯牙形態異常、爪の栄養障害がみられる。網膜血管新生を示す例がみられ、その場合は網膜剥離のリスクが増す。神経学的所見としては、てんかん発作、知的障害、発達遅滞などがみられることがある。
診断・検査
発端者におけるIPの診断は、1つ以上の大基準(特徴的皮膚病変)を有することで確定する。臨床症候だけでは断定しにくいような場合、女性発端者の場合はIKBKGの病的バリアントのヘテロ接合、男性発端者の場合は同ヘミ接合を同定することで、診断が確定する。
臨床的マネジメント
症状に対する治療:
水疱ならびに皮膚の感染に関しては標準治療を行う。小児歯科医の手で歯の管理を行う。必要に応じ、小児期にデンタルインプラントを行う。歯の異常のため、咀嚼や発音に問題が生じるようであれば、言語治療士や小児栄養士による管理を行う。網膜血管新生に対しては、網膜剥離のリスクを低減するための凍結治療やレーザー光凝固術を行う。てんかん発作・痙縮・局在病変の管理については、小児神経内科医への紹介を行う。機能的脳神経異常や網膜血管新生については、脳MRIを行う。発達遅滞に関しては、必要に応じ、発達プログラムや特別支援教育を行う。
二次的合併症の予防 :
皮膚の感染リスクを低減するための標準治療を行う。視力低下や斜視が現れたり、頭部外傷があったりした場合は、網膜剥離に関する評価を行う。
定期的サーベイランス :
眼の評価は、生後4ヵ月までは毎月、4ヵ月から1歳までは3ヵ月ごと、1歳から3歳までは6ヵ月ごと、3歳以降は年に1度の頻度で行う。神経機能の評価は、小児科医、小児神経内科医、発達小児科医への通常の受診の際に行う。歯については、小児歯科医ないし歯科医による通常の評価を行う。
リスクを有する血縁者の評価:
身体の診査と網膜の診査により罹患血縁者を特定することで、眼科スクリーニング検査の実施へとつなげることが可能になる。
遺伝カウンセリング
IPは、X連鎖性の遺伝形式をとる。罹患者の約65%は、de novoの病的バリアントに起因して生じた例である。ヘテロ接合の女性罹患者については、受胎の段階で子にIKBKGの病的バリアントを伝達するリスクが50%である。ただ、IKBKGの機能喪失型バリアントを有する男性胎児は、流産に至る。したがって、IP罹患者の母親から生まれた生産児については、非罹患女児が33%、罹患女児が33%、非罹患男児が33%の比率と予測される。現在までのところ、IP罹患男性はすべて、47,XXYの核型を有するか、もしくはIKBKGの病的バリアントの体細胞モザイクかのいずれかである。体細胞・生殖細胞系列両方についてIKBKGの病的バリアントをモザイクで有する男性の場合は、娘に対してはIKBKGの病的バリアントを伝達する(その病的バリアントを継承する女性は罹患者となる)可能性があるものの、息子に対してこれを伝達する可能性はない。家系内に存在する病的バリアントが同定されている場合は、高リスクの妊娠に備えた出生前検査や、着床前遺伝学的検査を行うことが可能である。
本疾患を示唆する所見
特徴的な口・顔・指の症候、多発性嚢胞腎、稗粒腫を有する女性については、OFD1を疑う必要がある。ただ、口・顔・指の症候は、他のタイプの口-顔-指症候群でもみられる。OFD1は、約50%の罹患者に多発性嚢胞腎がみられること、ならびに、複数の罹患者のいる家系についてはX連鎖性の継承パターンを示すことが特徴である。OFD1罹患者は、ほぼ全員が女性であるが、罹患男性の報告もいくつかみられる。ただ、こうしたものは、そのほとんどが、罹患女性が産んだ男性罹患胎児といった程度の記載にとどまっている。
皮膚、歯、毛髪、爪、眼、中枢神経系に特徴的臨床症候を有する例、ならびに以下に示すような家族歴を有する例については、色素失調症(IP)を疑う必要がある。
大基準(乳児期から成人期にかけて段階的に生じる皮膚病変)
- 紅斑
顔面を除く身体のあらゆる部位に、線状に分布する紅斑が現れ、次いでそれが水疱(小水疱)に変わる。水疱は数週のうちに消退するが、また新たなものが形成されることがある。紅斑が生じるのはステージⅠ(生後数週から24ヵ月;特に顕著なのは6ヵ月以前)である。
- 疣状病変
Blaschko線に沿った形で、主として四肢に疣状病変が現れる。これがステージⅡ(生後数週から24ヵ月)である。
- 線状ないし渦状の高色素性領域
Blaschko線に沿った形で、主として体幹にこれが現れ、思春期には褪色していく。これがステージⅢ(4ヵ月から16歳;稀に成人期まで続く例がある)である。
- 血色に欠けた色調、無毛、萎縮性の線条ないし領域
これがステージⅣ(思春期から成人期)である。
注:病変は上記のステージに従って出現することが多いものの、一時点で複数のタイプが混在するということが、どの時点についても起こりうる。ステージごとに、病変の出現部位が変わってくる可能性がある。
小基準
- 歯
部分性ないし全部性無歯症、小歯症(矮小歯)、形態異常歯
- 毛髪
脱毛症、羊毛状毛髪(艶がなく針金状のごわごわした毛髪)
- 爪
軽度の線状隆起形成ないし陥凹形成、鉤彎爪(肥厚し彎曲した爪)
- 網膜
周辺部の血管新生
- 家族歴
X連鎖性に一致した家族歴、ないし複数の流産歴
注:(1)小基準は、臨床診断を裏打ちするものとなる。
(2)Minićら[2014]は、小基準として、中枢神経系、口蓋、乳頭/乳房の形態異常を加えることを提唱している。
診断の確定
発端者におけるIPの診断は、1つ以上の大基準が存在することで確定する。臨床症候だけで断定しにくいような場合は、分子遺伝学的検査で次のバリアントの1つを同定することで、IPの診断を確定させることができる。
- 女性発端者におけるIKBKGのヘテロ接合性の病的(またはおそらく病的)のバリアント
- 男性発端者におけるIKBKGのヘミ接合性の病的バリアント
- 男性発端者におけるIKBKGの病的バリアントのモザイク(表1を参照)
注:(1)アメリカ臨床遺伝ゲノム学会(ACMG)/分子病理学会(AMP)のバリアントの解釈に関するガイドラインでは、「病的(pathogenic)」のバリアントと「おそらく病的(likelypathogenic)」のバリアントとは臨床の場では同義であり、ともに診断に供しうるものであると同時に、臨床的な意思決定に使用しうるものとされている[Richardsら2015]。本セクションで「病的バリアント」と言うとき、それは、あらゆるlikelypathogenicのバリアントまでを包含するものと理解されたい。
(2)意義不明のバリアントが同定された場合、それは、本疾患の診断を確定するものでも否定するものでもない。
分子遺伝学的検査として最も有効なものは、単一遺伝子検査である。
単一遺伝子検査
高頻度にみられる11.7kbにわたるIKBKGの欠失を標的にした解析をそれ単独で最初に行うか、もしくはIKBKGのシークエンス解析と並行して行い、そこで何の病的バリアントも検出されなかった場合は、次に遺伝子標的型欠失/重複解析を行うようにする。
注:高度な相同性を有する偽遺伝子IKBKGP1が存在するため、IKBKGの解析はより複雑なものになる。偽遺伝子に関する説明はここをクリック。
注:罹患男性については、体細胞モザイクのため、結果的にIKBKGの機能喪失型病的バリアントが検出されない結果になる可能性が考えられる。そのため、血液サンプルを用いた分子遺伝学的検査で病的バリアントが検出されなかった場合は、これに続いて組織サンプル(例えば、病変部の皮膚や精子)の分子遺伝学的検査を行うといったことが必要になる可能性がある。
マルチ遺伝子パネル
IKBKGその他の注目遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネルも検討に値する。
注:(1)パネルで用いられる手法としては、配列解析、欠失/重複解析、その他の非配列ベースの検査などがある。IKBKGの場合は、IKBKGP1が存在するため、次世代シーケンサー以外の手法を用いる必要がある。
(2)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(3)マルチ遺伝子パネルによっては、このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。したがって、どのマルチ遺伝子パネルを用いれば、現状の表現型と無関係な遺伝子の病的ないしおそらく病的のバリアントの検出を抑えつつ、問題にしている疾患の遺伝学的原因を特定できる可能性が高いかについて、臨床医の側であらかじめ検討しておく必要がある。
(4)検査機関によっては、パネルの内容がその機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(5)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。遺伝子検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
表1:色素失調症で用いられる分子遺伝学的検査
| 遺伝子1 | 手法 | その手法で病的バリアント2が検出される発端者の割合 | |
|---|---|---|---|
| 女性 | 男性 | ||
| IKBKG | 高頻度にみられる約11.7kbの欠失(c.399-?_1260+?del)を対象とした標的型解析 | 約65%3 | 3/18(16%)4 |
| 配列解析5 | 約8.6%4 | 2人6 | |
| 遺伝子標的型欠失/重複解析7 | 約4%8 | 報告例なし | |
| 不明9 | 検査対象外 | ||
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- この遺伝子で検出されているバリアントに関する情報については、「分子遺伝学」の項を参照のこと。
- Fuscoら[2008]
- 高頻度にみられる11.7kbの欠失を体細胞モザイクで有するIPの男性18人中の3人。
- 配列解析を行うことで、benign、likelybenign、意義不明、likelypathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、ミスセンス・ナンセンス・スプライス部位バリアントと遺伝子内小欠失/挿入などがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
- Fuscoら[2017]は、c.394C>Tの病的バリアントを体細胞モザイクで有する1男性例を報告している。Changら[2008]は、c.1167dupC(c.1167insCと表記されることもある)の病的バリアントを有する1男性例を報告している。この例は、HED-ID(乏汗性外胚葉異形成-免疫不全症)とIPの皮膚症候とを併せもつ唯一の男性例である(「遺伝学的に関連のある疾患」の項を参照)。
- 遺伝子標的型欠失/重複解析では、座位特異的な欠失や重複が検出される。具体的手法としては、定量的PCR、ロングレンジPCR、サザンブロッティングなど、さまざまなものがある。アッセイデザインは、IKBKGの偽遺伝子の存在を念頭に置いたものとする必要がある。
- IKBKGの座位について部分的なコピー数の増減を調べることを目的とした定量的PCR解析で、IKBKG、IKBKG偽遺伝子のみならず隣接遺伝子であるG6PDまでをも含んだ欠失に至るような組換え異常に起因して生じた病的バリアントが明らかになっている[Fuscoら2012,Conteら2014]。
- 今のところ、IPの原因となる別の座位が存在するというデータは存在しないものの、IKBKG相当座位の病的バリアントが検出されないIP罹患者が4.7%存在する。そうした例がある以上、座位異質性が存在する可能性も排除できない[Fuscoら2014]。
皮膚生検による病理組織検査
分子遺伝学的検査でIKBKGの病的バリアントが検出されない例については、皮膚生検を行って病理組織検査も検討対象になりうる。
- 罹患女性
特徴的な臨床症候を有する女性において、皮膚生検の組織診で好酸球浸潤や細胞外メラニン顆粒が確認される場合は、IPの診断の示唆ないし裏打ちになりうる。ただ、分子遺伝学的検査が広く行われ、その感度も上がっている今となっては、こうした検査が必要になるようなことはめったにない(表1参照)。それでも、分子遺伝学的検査で病的バリアントの同定に至らなかった境界線上の女性、あるいは疑いの余地のある所見を呈する女性について診断を補強していく上では、皮膚の生検が有用であるようなこともある。
- 罹患男性
皮膚生検の組織診は、むしろ男性例、特にモザイクの可能性が考えられる男性例で有用である。通常の病理検査であれば、分子遺伝学的検査用に採取した生検試料をそのまま使用して行うことも可能であろう。
臨床的特徴
臨床像
色素失調症(IP)は、皮膚、ならびにその付属器、眼、中枢神経系(CNS)の疾患であり、主として女性、時に男性に生じる。
臨床診断と分子診断の両方で診断が確定したIP罹患者のコホートの報告として最も大規模なものは、Fuscoら[2014]によるものである。
皮膚
図1、図2、図3、図4を参照されたい。IPの皮膚症候は、段階別に順を追って進む形の消長を示す。各ステージの始期と継続期間は症例によって異なり、必ずしも全例が4ステージすべてを通過するわけではない。各ステージを分ける基準は皮膚異常の様相であるが、これはBlaschko線という名で知られる胚・胎児期の皮膚の発生線に沿う形で現れる(図3参照)。Blaschko線は、胚発生期における細胞移動ないし成長経路に一致した形で現れる線である。デルマトームと同様、その走行は四肢では線状、体幹では円周状である。ただ、デルマトームと異なりBlaschko線は、神経の走行パターンや脊髄レベルに対応した形で現れるわけではない。
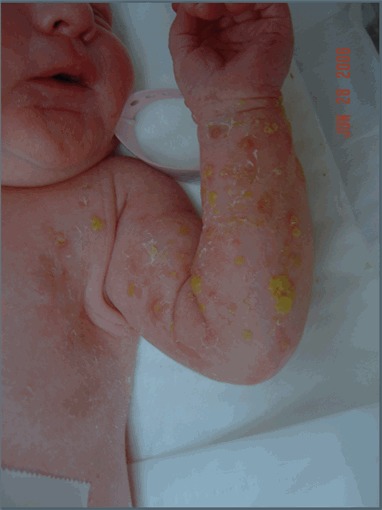
図1:ステージIのIP罹患女児
水疱は必ずしも線状に現れるわけではないことに注目されたい。
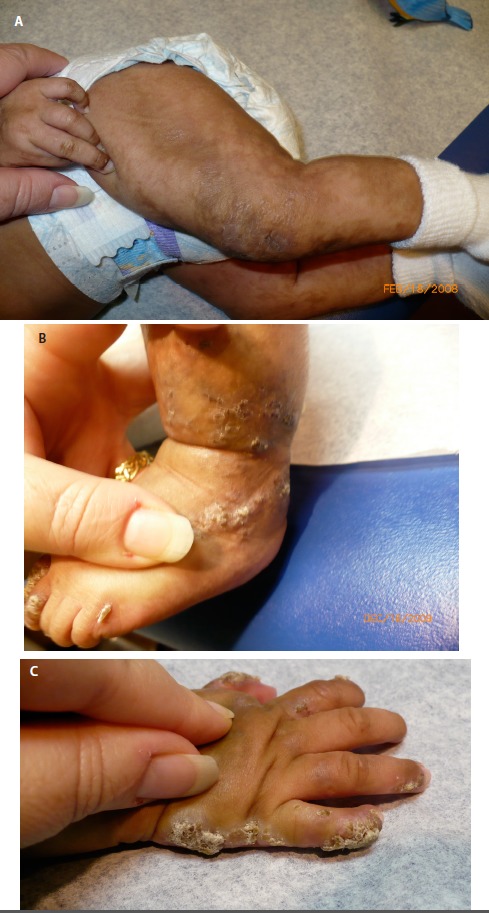
図2:ステージIIのIP罹患女児
疣状発疹期。疣状発疹は、必ずしもステージⅠと同じ部位に現れるわけではない。
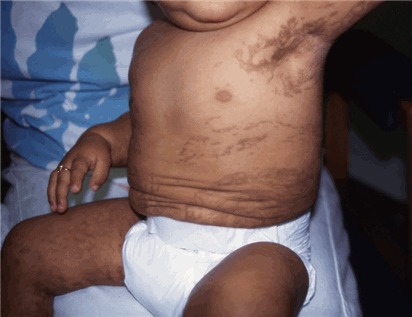
図3:ステージIIIの皮膚病変を呈するIP罹患女児
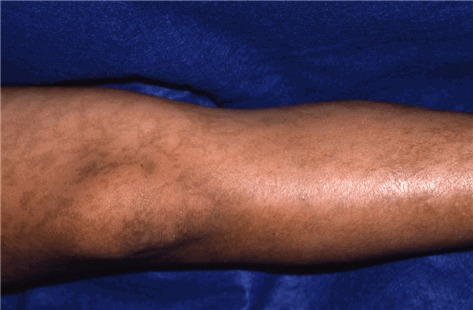
図4:網状の色素沈着パターンを示す成人例
- ステージⅠ―水疱期
水疱期は、水疱様の膨隆(図1)が生じることを特徴とする。これは四肢では線状、体幹では円周状に現れる。紅斑様であることも、感染巣様に見えるようなこともある。ステージⅠの始まりは生後6週から8週であるが、出生時にすでに認められることもある。ステージ1の水疱は、通常18ヵ月までに消退するものの、5歳の段階ですでにステージⅣの疹がみられた女児の1例で、水疱も同時にみられたとする報告が存在する[Darné&Carmichael2007]。
- ステージⅡ―疣状発疹期
疣状期は、肥厚性の疣状疹を特徴とする。これは、四肢では線状、体幹では円周状に現れる(図2)。ステージⅡの始まりは生後数ヵ月である。時に出生時にみられることがあるものの、通常はステージⅠが消退を始める時期に生じ始める。ステージⅡの持続期間は、通常2,3ヵ月間であるが、中には数年続く例もみられる。ステージⅡでは、爪ジストロフィーや歯の萌出様相の異常が明らかになることもある。
- ステージⅢ―色素沈着期
色素沈着期は、斑状でスレート様の灰色ないし茶色の色素沈着を特徴とする。これは、Blaschko線に沿った「マーブルケーキ」状ないし渦状パターン、すなわち、体幹では円周状、四肢では線状のパターンを示す(図3参照)。IPにおいては、この色素沈着期が最も特徴的なステージである。ただ、必ずしも広範囲にわたる色素沈着がすべての女性に生じるわけではなく、ごく限られた部位にのみ生じることがありうる。最も高頻度にみられる部位は、鼠径部と腋窩である。こうした特徴的パターンを特定するためには、全身の皮膚をくまなく診査する必要がある。色素沈着が現れ始めるのは生後6ヵ月から1歳までの間で、通常はステージⅡが消退し始める時期にあたる。こうした色素沈着が出生時からみられることは決してない。ステージⅢは、時に成人期まで続くことがある。ただ通常は、ティーンの時期ないし成人初期には色素沈着が消退し始める(図4参照)。こうした色素変化は、線状に現れることもあれば、渦状、網状にみられることもある。30歳代以降の女性の場合は、IP関連の皮膚変化が一切みられないこともある。
- ステージⅣ―消退期
消退期の特徴は、線状色素消退ないし脱毛が、四肢や頭皮を中心に現れることである。ステージⅣの定義については、まだ意見の一致をみていない。真性の色素消退がみられず、毛髪や皮腺の消失のみがみられる場合もある。ステージⅠ-Ⅲと同様、出現パターンはBlaschko線に沿った形をとる。ステージⅣは、必ずしもすべての罹患者に現れるわけではない。現れる場合は、色素沈着が消失した後の時期からこのステージが始まる。
毛髪
脱毛は、頭皮に加えて体幹や四肢にも生じることがある。頭皮におけるパッチ状の脱毛部位はステージⅠに水疱がみられた部位の瘢痕部位に一致して現れることがある一方で、ステージⅠやⅡで頭皮に病変がみられなかった罹患者に出現することもある。脱毛は、ステージⅣの皮膚変化の一部としての皮膚の色素消退部位に一致して出現する。幼児期には、頭髪が細くなったり疎になったりすることがある。頭頂部の「羊毛状毛髪母斑」部位では、しばしば、艶がなく針金状でごわごわした毛髪となる。脱毛部位の面積は非常に小さいことがあり、本人も気づいておらず、周りの頭髪で隠されているような場合は、発見が難しいこともある。睫毛や眉毛が疎であったとする報告もみられる。
歯
歯の異常としては、無歯症(歯数の減少)、小歯症(矮小歯)、形態異常歯(例えば円錐歯や副咬頭)、萌出遅延、埋伏歯などがある。エナメル質や歯の強度は正常である。IP罹患者で報告されている歯の異常には大きな幅がみられ、歯の形や数に関するあらゆる異常が起こりうると言っても過言ではない。
爪
爪にジストロフィー(線条隆起、陥凹、脆弱化)がみられることがある。こうした変化は、真菌感染を起こした爪と類似することが多い。爪のジストロフィーは、ステージⅡで最も多くみられる。こうした爪の変化は一時的のこともあるが、爪に消えることのない1本の縦線が、罹患者の28%にみられたとする研究が存在する[Phanら2005]。
眼
IP罹患者は眼の異常に関して高リスク(20%―77%)である。
- 網膜
網膜の血管過剰増生が最も多くみられる。治療を行うことなく放置すると、網膜剥離に至る。網膜剥離に関して最も高リスクなのは乳幼児期である。6歳を過ぎて生じることはほとんどない。こうした変化は、散瞳下での倒像眼底検査で可視化が可能である。
- その他の眼の症候
その他の眼の症候としては、斜視、白内障、視神経萎縮、網膜色素異常、小眼球などがある[Meuwissen&Mancini2012,Fuscoら2014]。
中枢神経系
てんかん発作、知的障害その他の中枢神経系の異常がIP罹患者の約30%で報告されている[Minićら2014]。ただ、神経認知機能障害を伴わない軽症の罹患者については、医学的注目を集めるに至らないと思われることから、認知機能障害の実際の発生頻度はよくわかっていない[Phanら2005]。認知機能障害は、家族例より孤発例でより高頻度にみられるが、これはおそらく、家族性の場合は結果的に軽症の血縁者もIPとして同定されやすいためと思われる。男性のIP罹患者は、女性のIP罹患者より神経学的異常を有する例が多い。一般論として、IP罹患者にみられる神経学的異常は、その背後にある中枢神経系の血管障害の反映であるように思われる[Meuwissen&Mancini2012]。
- てんかん発作
IPにみられるてんかん発作は、生涯にたった一度だけの発作から、慢性のてんかんに至るまで、幅がみられる。卒中は、大脳内のいずれの部位にも生じる可能性があるため、発作の種類はさまざまである。Meuwissen&Mancini[2012]は、神経認知機能障害を有するIP罹患者の報告の中で、詳細な記載がなされているものについてレビューを行っており、その報告において、各種発作の中で焦点性間代発作が最も多くみられたと述べている。認知機能障害を有するIP罹患者の約25%(IP罹患者全体の約7%)が、てんかん発作を1回以上経験していた。てんかん発作は、その大多数が生後1年以内に発生する(報告中で初発年齢が記載されている35人中32人)。再発の有無に言及している報告では、発作の出現が1回のみであった例が、25人中14人に上っていた[Meuwissen&Mancini2012]。
- 知能
IPの認知機能を評価した研究は多くない。結果には幅がみられ、正常とする報告もみられる。重度の知的障害の報告は多くない。男性では、47,XXYの核型を有する例を含むだけに、IPの知能面での表現型がより複雑になっている可能性がある。Pizzamiglioら[2014]は、認知機能の評価を行った女性IP罹患者10人について報告している。10人中7人については、計算/数学的推論ならびに読字能力に障害がみられたものの、書字能力に問題はみられなかった。この評価を受けて、IPの症候の1つに「学習障害」を加えることが可能となった。2017年に発表された追跡評価の論文[Pizzamiglioら2017]によると、14人中9人が正常な発達を示す一方、5人には、軽度から重度までの知的障害がみられたという。
・脳奇形
原発性の脳奇形は稀である。これまでに、後頭部脳瘤を伴う脳梁無形成[Demirelら2009]、多小脳回[Godambeら2005]、異所性灰白質[Mangano&Barbagallo1993]が、それぞれ別の罹患者で報告されている。
IKBKGの病的バリアントにより微小血管に異常が生じる可能性のあることが明らかになったことで、中枢神経系の機能障害が、血管障害の結果として生じた一過性脳虚血発作や本格的な出血性卒中に起因する二次性のものであるという考え方が裏打ちされる結果になった[Fiorilloら2003,Hennelら2003,Shahら2003]。神経血管異常は、満1歳に達するまでの間に最も多くみられ、それ以降は数えるほどしかなく、4歳以降の報告例は3例に過ぎない[Meuwissen&Mancini2012]。
てんかん発作を中心とした認知機能障害を有するIP罹患者について脳のMRIを行ったところ、43人中27人に脳室周囲白質軟化がみられた。また、皮質下の白質の変化も多くみられた。その結果としての嚢胞性変化を示した例もいくつかみられた。髄鞘形成遅延、脳室拡大の報告もみられる[Meuwissen&Mancini2012]。
- 痙性麻痺
痙性麻痺の発生頻度については、よくわかっていない。古い文献に記された内容は、解釈が難しいように思われる。IPでみられるその他の神経学的異常と同様、痙性麻痺のリスクや重症度についても、中枢神経系の血管障害次第という側面があるように思われる。
乳房
乳房無形成から副乳に至るまで、乳房組織の異常には大きな幅がみられるものの、いずれにしても一般集団よりは高頻度に生じる[Minićら2014]。報告例は思春期前の症例に偏っているため、認知されている乳房異常の発現頻度は、過少に評価されている可能性がある。
その他
- 白血球増多症
好酸球が最大で65%を占める白血球増多症が、ステージⅠ、ステージⅡを中心として生じる可能性がある。白血球増多症を引き起こす原因は特定されていない。Zilberman-Rudenkoら[2016]は、IKBKGの病的バリアントによりNF-κBの抑制が阻害され、炎症反応の応答性亢進が引き起こされることを報告している。好酸球増多症は、いずれの臨床症候とも一貫した関連性をもたず、通常は自然消退に至る。
- 原発性肺高血圧
一部の罹患者で原発性肺高血圧が報告されている。これはおそらく、血管症に起因するものと考えられる[Alshenqitiら2017]。
IP罹患男性について
IPは、「男性致死性」疾患とされているが、詳しく調べられて報告された男性例も実際に存在する。毎年数例が報告され、文献のレビューも散発的ながら登場する。男性が生存に至るメカニズムとしては次の2通りがある。
- 47,XXYの核型をもつこと。IP罹患男性の7%がこれであると推定されている[Pachecoら2006]。
- 体細胞モザイク
- X染色体とY染色体のプローブを用いた間期核FISH法を用いて初めてモザイクであることが証明されるに至った46,XY/47,XXYの低レベルモザイクの1男性例が報告されている[Francoら2006]。この罹患児のIKBKGの病的バリアントは、実証可能な形では確認できなかった。
- IKBKGの機能喪失型病的バリアントの低レベルモザイクが2人の男性で同定されている。線維芽細胞や精子のDNAにおいて、異常細胞が最も高い割合でみられた[Fuscoら2017]。
- 男性例の中にも「分節型」IP(四肢の1つにのみ病変が現れる状態)を示す例が確認されている。これは、体細胞モザイクに一致する所見である。
IPで男性が致死性となる理由は、IKBKGのバリアントを有するX染色体を継承した男性受精卵の場合、生存に必要な正常タンパク質が欠損することにある。男性が致死性となるメカニズムの詳細は不明である[Hatchwell1996]が、マウスモデルでは、肝不全が一定の関与を示すことが示唆されている[Rudolphら2000]。
軽症型IPを引き起こす病的バリアントは、男性においては常に免疫不全を伴う形で現れ、X連鎖性乏汗性外胚葉異形成-免疫不全症(HED-ID)の名で知られている[Fuscoら2008]。HED-IDで、IPの臨床症候を併せもつ男性が1例のみ、IKBKGに現れたc.1167dupCのバリアントに伴って生じた例として報告されている[Changら2008]。
平均寿命
新生児期や乳児期に重大な合併症がなかった例については、平均寿命は通常と変わらないと考えられている。
生殖適応度
IPの女性は、妊娠喪失に関して高リスク状態にある。それは、男性胎児の生存率の低さに伴うものと推察される。IP罹患女性が複数回にわたって流産を経験することはよくあることで、その多くが妊娠3ヵ月目か4ヵ月目頃に生じる。その点を除いては、妊孕性に問題はみられない模様で、非罹患胎児を受胎した場合は、妊娠・分娩に際して特段の合併症は生じないものと考えられている。
遺伝型-表現型相関
病的バリアントの中の1つのグループ(主としてエクソン10に生じたバリアント)については、NF-κBシグナル伝達が減弱はされるものの、完全喪失にまでは至らない。そのため、女児については軽症型IPの表現型が現れ、男性胎児についても流産のリスクが低くなる。さらに、こうしたバリアントの大部分(ミスセンスバリアント,フレームシフトを生じる1塩基の挿入/欠失,ナンセンスバリアントなど)においては、男性は、乏汗性外胚葉異形成-免疫不全症(HED-ID)や、免疫不全・大理石骨病・リンパ浮腫を伴う無汗性外胚葉異形成症(OLEDAID)(訳注:「2019年版骨系統疾患国際分類の和訳」では「外胚葉異形成と免疫不全を伴う大理石骨病」となっている)を有しつつも、生存可能である(「遺伝学的に関連のある疾患」の項を参照)。
浸透率
色素失調症は高い浸透率を示す。IP罹患者の大多数については、生後数ヵ月のうちに表現型が表に現れる。
しかし、表現度については非常に大きな幅がみられる。加えて、皮膚症候は経時的に消失していくことがあり、また年齢とともに他の皮膚疾患と区別できなくなることもある。さらに、歯、毛髪、爪の異常は美容的対応が可能な部分でもあり、成人女性の場合、臨床診査だけでは診断上の症候を見つけることが難しいこともありうる。
疾患名について
X染色体の構造異常を有する罹患者の中に、その異常がIKBKGの座位(Xq28)とは無関係な部位に生じているにもかかわらず、渦状の色素沈着を呈する例がみられる。こうした観察結果から、Xp11が原因座位と推察される独立疾患としての「色素失調症Ⅰ型」なる病名が、かつてこれに割り当てられたことがある。しかし詳細に調べたところ、Xp11との連鎖や表現型の一貫性は確認されなかった。そのため、現在では、これに「色素失調症Ⅰ型」という病名を当てることは適切ではないと考えられている[Happle1998]。
発生頻度
IPを有する女性は、これまでに2,000例以上報告されている[Minićら2014]。公衆衛生先天異常監視システムのデータでは、IPの出生時発生頻度について、1,000,000人あたり0.6-0.7人となっている[Orphanet,TexasBirthDefectsRegistry,未公表データ]。2023年、Orphanetは、EU域内における出生時発生頻度について、100,000人あたり1.2人との推定値を出している。またHerlinら[2024]は、いくつかのデンマーク国営医学レジストリ・データベースを用いた推定値として、100,000人あたり2.37人(95%信頼区間:1.74-3.25人)、すなわち、42,194人に1人という数字を出している。これは、従来の推定値の約2倍の高さである。女性:男性比は20:1である[OrphanetReportSeries2017]。
遺伝学的に関連のある疾患(同一アレル疾患)
- 乏汗性外胚葉異形成-免疫不全症(HED-ID)(OMIM300291)
- 免疫不全・大理石骨病・リンパ浮腫を伴う無汗性外胚葉異形成症(OLEDAID)(OMIM300301)
この2疾患は男性のみが罹患する。両疾患は、IKBKG内に生じた主としてミスセンスバリアント(ただし、インフレーム欠失、フレームシフトバリアント、スプライス部位バリアント、その他の病的バリアントも知られている)に起因して生じ、これにより核内因子κB(NF-κB)シグナル伝達の減弱(阻害ではない)を招いた結果として生じるものである。HED-IDに関しては、これに関与するIKBKGの病的バリアントのリストが報告されている[Fuscoら2008]。
注:HED-IDやOLEDAIDは、X連鎖型の乏汗性外胚葉異形成症とは明確に別の疾患である。後者はEDAの病的バリアントないし欠失によって生じる疾患である(「乏汗性外胚葉異形成症」のGeneReviewを参照)。
- 免疫不全症33/X連鎖性抗酸菌症(IMD33,AMCBX1)(OMIM300636)
これは、初期には化膿性細菌、その後、抗酸菌に対して感受性を示す複合免疫不全症として現れる。男性において、IKBKGの特定の病的バリアントに起因して生じるものとして報告されている。
鑑別診断
骨格病変(神経障害に起因して二次性に現れるものを除く)、重度の神経障害、重度の脱毛、非定型色素沈着、重度の色素消退がみられる場合は、色素失調症(IP)以外の疾患を検討する必要がある。通常、IPは身体の非対称を伴わないが、これまでにIP罹患者で上肢遠位末端の横断型欠損を示した1例が報告されている[Hayesら2005]。
IPの皮膚症候に関する鑑別診断は、ステージごとに変わってくる。IP罹患児は感染症にも罹りやすい状態になっているため、IPであるか否かにかかわらず、感染症の所見に関しては、それに従った評価を行っていく必要がある。
- ステージⅠ―水疱期
以下の疾患を念頭に置く必要がある。先天性単純ヘルペス,水痘、ブドウ球菌ないし連鎖球菌を原因とする水疱性膿痂疹,(重症例では)表皮水疱症(「栄養障害型表皮水疱症」,「単純性表皮水疱症」のGeneReviewを参照)。感染の際は通常、発熱、全身毒性の症候等、他の炎症のサインが現れる。感染の診断は、病変部の組織を採取して培養することで行われる。軽微な外傷の後に水疱が形成されるということであれば、それは表皮水疱症の特徴である。診断の確定は、皮膚生検の解析、透過電子顕微鏡検査、免疫蛍光抗体/抗原マッピング、分子遺伝学的検査により行われる。
- ステージⅡ―疣状発疹期
この時期の症候が他の疾患と混同される可能性は低いように思われるが、軽度のIP罹患者の皮膚症候については、単純な疣や伝染性軟属腫と類似することがある。一定のパターンをもって多数出現するような場合は、疣や伝染性軟属腫ではなくIPである可能性が高い。IPの病変1個のみを取り上げて、これを疣と鑑別することは、生検を行わない限り困難な場合もある。
- ステージⅢ―色素沈着期
不規則な形で皮膚の色素沈着が生じたり、Blaschko線に沿った形でその他の異常が現れたりするするあらゆる疾患が鑑別の対象となる。線状、渦状の色素変化は、モザイク型の染色体異常でしばしばみられる所見である。ただ、モザイク型染色体異常の場合は、脳奇形をはじめとする先天奇形や知的障害が多くみられ、色素異常も発疹に続いて現れるというパターンではなく出生時からみられる。こうした例については、血液サンプルないし皮膚(線維芽細胞)サンプルを用いた通常の核型解析を検討すべきである。伊藤白斑(OMIM300337)は、モザイク型染色体異常に伴って現れる1つの表現型である。もう1つの重要な鑑別点は、IP罹患者の場合、異常なのは高色素領域のほうであるのに対し、伊藤白斑の場合は低色素領域のほうが異常だという点である。モザイク型染色体異常を有する罹患者については、どのレベルの色素沈着が、その人にとって「正常」なのかの判定が難しいことがしばしばある。
- ステージⅣ―消退期
病変が終息しつつある部位の皮膚は、瘢痕、(限局性の脱毛を伴う)白斑、その他の低色素と局所の脱毛を呈するあらゆる状況と類似性を示す。鑑別は主として病歴に基づいて行われる。白斑の場合は進行性で、低色素領域の周りを高色素領域が取り囲む形となる。また、IPの他のステージに現れるような病変が白斑に先行して現れるといったことはなく、皮膚以外の症候を伴うようなこともない。限局性白皮症(OMIM172800)は、常染色体顕性遺伝の形式をとる低色素疾患で、症候は皮膚のみに限られ、ふつうは出生の段階ですでに存在し、その後進行はしない。
IPでみられるその他の症候について、鑑別を要する疾患には以下のようなものがある。
- Naegeli症候群(OMIM161000)
Naegeli症候群は、皮膚ならびに皮膚由来組織に症候が現れる稀な常染色体顕性遺伝疾患で、IPとの類似性を示す一方で、多汗症や手掌・足底の点状過角化症といった症候を併せて呈する。IPと異なり、Naegeli症候群の場合は、皮膚の症候が段階を追って進んでいくことはない。Naegeli症候群はきわめて稀な疾患であり、線状の疣状病変を呈する場合は、IPである可能性のほうが高い。Naegeli症候群は、KRT14の病的バリアントに起因して生じる疾患である。
- 網膜の血管新生
網膜の血管新生は、未熟児網膜症や家族性滲出性硝子体網膜症においてもみられる。そして、Norrie病スペクトラム(GeneReviewsの「NDP-relatedRetinopathies」を参照)の部分症候としてX連鎖潜性遺伝で継承されたり、常染色体顕性の遺伝形式(GeneReviewsの「OMIMPhenotypicSeries:Exudativevitreoretinopathy」を参照)をとったりする可能性がある。これらの疾患においては、皮膚症候が現れることはない。
臨床的マネジメント
最初の診断に続いて行う評価
色素失調症(IP)と診断された罹患者については、疾患の範囲やニーズを把握するため、もしもまだ行っていないならば、以下の評価を行うことが推奨される。
- 症候の有無ならびに範囲を特定することを目的として、特に皮膚、毛髪、爪、神経系に重点を置いて行う身体診察
- 臨床遺伝医ないし遺伝カウンセラーとの面談
- 重度の皮膚症候を有する罹患者については、その管理を目的とした小児皮膚科医の診察
- 網膜の血管新生の有無を調べるための、IPないし網膜疾患に精通した眼科医による迅速な検査
- 脳のMRI検査、てんかん発作その他の神経系の異常ないし網膜の血管新生がみられる場合は、脳波検査を目的に神経内科医への紹介
- 磁気共鳴血管造影。神経障害の状態が卒中様パターンに一致する場合、脳血管障害部位の特定にこれが有用な可能性がある。
- 発達遅滞が明らかな場合は発達評価
- 肺高血圧を有する新生児の管理を目的とした小児循環器内科医の治療への参画
症候に対する治療
治療の内容は以下の通りである。
- 水疱については、標準的な手法(すなわち、切開しない,外傷を避ける)で治療を行う。不快症状の緩和には局所療法(例えば、投薬,オートミール浴)を行う。重度の場合は皮膚科での管理が有効な場合もある。
- 他の蜂窩織炎と同様の感染症治療を行う。
- 生後6ヵ月、あるいは歯の萌出開始のいずれか早いほうの段階で、小児歯科医への紹介を行う。デンタルインプラントについては、7歳で行われた例もある(IPと同様の歯科的問題を有する外胚葉異形成の子ども;GeneReviewsの「Hypohidroticectodermaldysplasia」を参照(GRJに和訳あり))。
- 乳歯の萌出遅延その他の障害に伴って咀嚼や言語の発達が障害されている場合は、言語治療士や小児栄養士への紹介を行う。
- 網膜剥離を引き起こす可能性のある網膜の血管新生に対しては、凍結治療とレーザー光凝固術を行う。
- 網膜剥離に対しては、標準治療を行う。
- てんかん発作の治療が必要な場合、ならびに痙縮、局在病変、網膜の血管過剰増生がみられる場合は、小児神経内科医への紹介を行う。
- 機能性の神経異常や網膜の血管過新生を有する小児については、例外なく脳のMRIを行う。
- 発達遅滞に対しては、必要に応じて適切な発達刺激策や特別支援教育を行う。
- 新生児肺高血圧に対しては、標準治療を行う。
二次的合併症の予防
新生児期は、標準的手法、すなわち密封状態にある水疱を破裂させない、治癒過程にある患部を清潔に保つ、過度の炎症がみられないか、全身に影響が及んでいないかといった点のモニタリングを慎重に行うといった、水疱の感染リスク低減を図る管理が中心となる。
特に7歳未満の子どもについては網膜剥離の危険性がある点を両親に説明し、視力に何らかの変化がみられるとき、後天性斜視の徴候がみられるときは、直ちに受診させる。頭部外傷は網膜剥離の誘因になるため、頭部外傷を起こした際の評価項目として、詳細な眼の検査を含める必要がある。ただし現時点では、コンタクトスポーツを避けることに関し、特別な勧告は出されていない。
定期的サーベイランス
眼科検診については、今のところ確立したスケジュールというものは存在しないものの、おおむね次のように行うことが提唱されている。
- 生後3,4ヵ月までは毎月
- 生後4ヵ月から1歳までは3ヵ月ごと
- 1歳から3歳までは6ヵ月ごと
- 3歳以降は年に1度
神経発達に関しては、小児科医、小児神経内科医、発達小児科医のいずれかの手で、来院ごとに評価していく必要がある。
小児歯科医や歯科医による評価も、継続的に行っていくことが望ましい。
リスクを有する血縁者の評価
速やかに治療を開始したり、予防的措置(定期的な眼科検診)を講じたりすることで利益が得られる人を可能な限り早期に特定していくため、リスクを有する血縁者に対しては、発端者より年上であるか年下であるかを問わず、一見したところ無症候と思われる場合であっても、評価を行うべきである。
評価項目は以下の通りである。
- 家系内に存在する病的バリアントが既知の場合は、分子遺伝学的検査
- 家系内に存在する病的バリアントが未知の場合は、皮膚、歯、毛髪、爪、網膜の診査、神経学的評価を含む身体診察
遺伝カウンセリングを目的として、リスクを有する血族に対して行う検査関連の事項については、「遺伝カウンセリング」の項を参照のこと。
妊娠に関する管理
妊娠の際の健康管理や対応に関する全般的事項については、通常の場合と特に変わりがないことが多い。胎児の生存の如何に関連して生じる自然流産の比率は、一般集団より高いものの、妊娠損失に対する管理法については、標準的なやり方で行われる。網膜に問題を抱える女性については、網膜剥離を避ける意味合いから、体力の消耗を最小限に抑える、あるいはゼロにするための分娩管理を検討する必要がある。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「ClinicalTrials.gov」、ならびにヨーロッパの「EUClinicalTrialsRegister」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
色素失調症(IP)は、X連鎖性の遺伝形式をとる。
家族構成員のリスク
発端者の両親
女性発端者の親
- IP罹患女性は、IKBKGの病的バリアントを母親から継承する場合と、denovoの病的バリアントに起因する場合の2通りがある。
- Denovoの病的バリアントに起因してIPが生じる場合は、そのバリアントは父親由来のIKBKGアレルに生じたものであることが多い[Smahiら2000,Fuscoら2004]。
- IKBKGに生じるdenovoの病的バリアントが多くみられる。高頻度にみられる11.7kbの欠失を有するIP罹患女性91人中59人(65%)が、denovoの病的バリアントであった[Fuscoら2009]。
- 母親もIPの診断基準を満たしている場合、あるいは母親の第1度近親に別にもう1人罹患者がいる場合、母親はIKBKGの病的バリアントを有している。
- 発端者の有するIKBKGの病的バリアントがすでに判明している場合は、母親に対して分子遺伝学的検査を行うことが可能である。
注:表現度には大きな幅がみられるため、すでに成人に達した女性については、自分が子ども時代に軽度の症候を有していたことを忘れてしまっており、成人した今となっては、もはやすぐにそれとわかる身体的症候は何も残っていないことがありうる。
男性発端者の親
- 接合後にIKBKGの病的バリアントのモザイクが生じたためにIP罹患者となった男性発端者については、その母親や父親が病的バリアントをそれぞれヘテロ接合、ヘミ接合で有している可能性はゼロである。
- 男性発端者が47,XXYの核型を有している場合、その母親はIKBKGの病的バリアントをヘテロで有している可能性があるため、母親の分子遺伝学的検査を行うことが望ましい[Kenwrickら2001]。
女性の発端者の同胞
同胞の有するリスクは、両親の遺伝学的状態によって変わってくる。
- 女性発端者の母親
- 女性発端者の母親が罹患者であった場合、受精の段階で同胞がIKBKGの病的バリアントを継承するリスクは50%である。ただしIKBKGの機能喪失型バリアントを有する男性胎児は、その大多数が流産に至る。そのため、分娩の段階における比率は、おおむね、非罹患女性が33%、罹患女性が33%、非罹患男性が33%という割合になる。
- 母親もIP罹患者で、有しているIKBKGの病的バリアントがタンパク質活性を低下(「喪失」ではなく)させるタイプである場合、男性胎児は出生時に免疫不全を伴う無汗性外胚葉異形成症(EDA-ID)を示しつつも生存する可能性が出てくる。
注:母親がIP罹患者で、高頻度にみられる11.7kbの欠失(これによりタンパク質の活性は完全に失われる)を有している場合であれば、EDA-IDの生産児が産まれることに関し、特段のリスク上昇はない。
- 女性発端者の父親
父親がIKBKGの病的バリアントを体細胞ならびに生殖細胞系列のモザイクで有していた場合、すべての女性同胞はそのバリアントを継承して罹患者となるリスクを有する。
男性同胞がバリアントを継承するリスクはない。IKBKGの機能喪失型バリアントを体細胞ならびに生殖細胞系列のモザイクで有する父親から娘へIPが伝達された例が、これまでに複数家系報告されている[Fuscoら2017]。
- 発端者が孤発例(家系内で1人だけの発生)で、IKBKGの病的バリアントが両親いずれの白血球DNAからも検出されない場合、同胞の有するリスクは一般集団の数字より若干高い程度(それでも1%未満)になる。それは父母のいずれかが生殖細胞系列モザイクの可能性が残っているからである[Fuscoら2017]。
男性の発端者の同胞
同胞のリスクは、母親の遺伝学的状況により変わってくる(「女性発端者の同胞」の項を参照)。
女性発端者の子(図5参照)
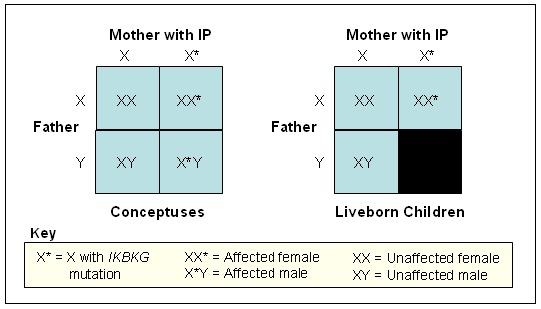
図5:胚の遺伝型と生産児の遺伝型との比較対照
- IP罹患女性の子のリスクを考える上では、妊娠期間中に罹患男性胎児が死亡に至ると思われる点を考慮する必要がある(図5)。
- 受精の段階でIKBKGの病的バリアントが伝達されるリスクは50%であるが、IKBKGの機能喪失型バリアントを有する男性胎児は、その大多数が流産に至る。そのため、分娩の段階で子に生じると予想される比率は、非罹患女児が33%、罹患女児が33%、非罹患男児が33%となる。
- IP罹患者である母親が、タンパク質の活性低下(活性喪失ではない)に至るIKBKGの病的バリアントを有している場合は、男性胎児が生存して、出生の段階でEDA-IDを示す可能性が出てくる。
注:母親が、高頻度にみられる11.7kbの欠失(これによりタンパク質の活性は完全に失われる)を有するIP罹患者である場合は、EDA-IDの生産男児が産まれることに関し、特段のリスク上昇はない。
男性発端者の子
- 現在までのところ、IP罹患男性は47,XXYの核型を有する例か、IKBKGの病的バリアントを体細胞モザイクで有する例かに限られている。
- 体細胞モザイクと同時に、生殖細胞系列モザイクも有する罹患男性については、IKBKGの病的バリアントを娘に伝達する可能性がある(病的バリアントを継承した娘はすべて罹患者となる)。息子については、病的バリアントを継承するリスクはない。
他の血縁者
- 発端者の片親がIKBKGの病的バリアントを有しているならば、その血縁者はすべて罹患者であるリスクがある。
- 発端者のIKBKGの病的バリアントが判明していないものの、同じ病的バリアントを共有する女性血縁者を同定したい場合には、X染色体不活化の偏りの状況を調べる検査が有用なことがある。
関連する遺伝カウンセリング上の諸事項
リスクのある血縁者に対して、早期診断、早期治療を目的として行う検査関連の情報については、「臨床的マネジメント」の中の「リスクを有する血縁者の評価」の項を参照されたい。
他の多くの遺伝性疾患の場合と同様、新生児におけるIPの診断の際に、家系内に存在していた遺伝性疾患の存在にそれまで気づいていなかった母親その他の血縁者に対して、評価や診断を行うことになりうる。母親その他の血縁者にとって、それは自身の健康状態を明らかにすることであり、同時に子のもつ病気の「責任」が自分にあるとの自責の念を生むことでもあるだけに、新生児におけるIPの診断は、難しさを伴うものになりうる。そうした問題も頭に入れながら努力していく必要があろう。
家族計画
- 遺伝学的リスクの確定や、出生前/着床前遺伝学的検査を受けるかどうかの話し合いに最も適しているのは、妊娠前の時期である。
- 罹患者である、IKBKGの病的バリアントを有している、有リスク状態にあるといった状況にある若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法や、遺伝子・病原メカニズム・疾患等に対するわれわれの理解は、将来より進歩していくことが予想される。そのため、分子遺伝学的な診断が確定していない(すなわち、原因となった病原メカニズムがわかっていない)発端者のDNAについては、保存しておくことを検討すべきである。詳細は、Huangら[2022]を参照されたい。
出生前検査ならびに着床前遺伝学的検査
家系内に存在するIKBKGの病的バリアントが同定されている場合は、出生前診断や着床前遺伝学的検査を行うことが可能である。
罹患男性と罹患女性とでは予後が大きく異なる。したがって、遺伝カウンセリングに正確を期す上では、胎児の核型を確定しておくことが不可欠である。
- 胎児の核型が46,XXであった場合は、両親に対し胎児がIPに罹患している可能性は50%であるとの説明を行う。
- 胎児の核型が46,XYであった場合は、カウンセリングに際して、罹患胎児については第1三半期を過ぎると流産に至るリスクが高いという説明を併せて行う。
- 胎児の核型が47,XXYであった場合は、IPは男性のほうが重度の表現型になるという説明、ならびにKlinefelter症候群に関する説明を併せて行う。
注:女性においてIPの表現型がより軽度な形で現れる一群の病的バリアント(主としてエクソン10に生じるバリアント)が知られており、その場合は、流産のリスクが低くなる(「遺伝型-表現型相関」の項を参照)。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- IncontinentiaPigmentiASSociazioneItaliana(I.P.ASS.I.)–Onlus
ViaAltair,5
00012GuidoniaMontecelio
Italy
Phone:393332089513
Email:presidenza@incontinentiapigmenti.it
www.incontinentiapigmenti.it - IncontinentiaPigmentiInternationalFoundation(IPIF)
30East72ndStreet
NewYorkNY10021
Phone:212-452-1231
Fax:212-452-1406
Email:ipif@ipif.org
www.ipif.org - NationalInstituteofNeurologicalDisordersandStroke(NINDS)
POBox5801
BethesdaMD20824
Phone:800-352-9424(toll-free);301-496-5751;301-468-5981(TTY)
Incontinentia Pigmenti Information Page - NationalLibraryofMedicineGeneticsHomeReference
Incontinentia pigmenti - IncontinentiaPigmentiGeneticBiobank(IPGB)
IGB-CNR
ViaP.Castellino,111
Naples80131
Italy
Phone:+390816132302
Fax:+390816132706
Email:incontinentia.pigmenti@igb.cnr.it
www.igb.cnr.it/ipgb
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:色素失調症:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specificデータベース | HGMG | ClinVar |
|---|---|---|---|---|---|
| IKBKG | Xq28 | 核内-kappa-B essential modulator | IKBKG @ LOVD IKBKGbase: Mutation registry for Nemo deficiency |
IKBKG | IKBKG |
データは、以下の標準資料から作成したものである。遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:色素失調症関連のOMIMエントリー(閲覧はすべてOMIMへ)
| 300248 | INHIBITOR OF NUCLEAR FACTOR KAPPA-B KINASE, REGULATORY SUBUNIT GAMMA; IKBKG |
| 308300 | INCONTINENTIA PIGMENTI; IP |
分子レベルの病原
IKBKG周辺のゲノム構成は複雑である。IKBKG(以前はNEMOと呼ばれていた)内には、MER67Bと呼ばれる870bpのダイレクトリピートが2つ存在し、1つはイントロン3内に、もう1つはIKBKGの下流に位置している(図6)。2つのMER67Bの中間部分に組換えが起こることで、IKBKGのエクソン4から10まで(以前はIKBKGP1と呼ばれていた)に欠失が生じる。これが、高頻度にみられる11.7kbの欠失である(表2)。この領域にあるこれ以外の複雑リピートエレメントに生じる再構成は良性のバリアントであり、一般集団内で反復性にみられる(1-2%の頻度と推定される)(図6)。
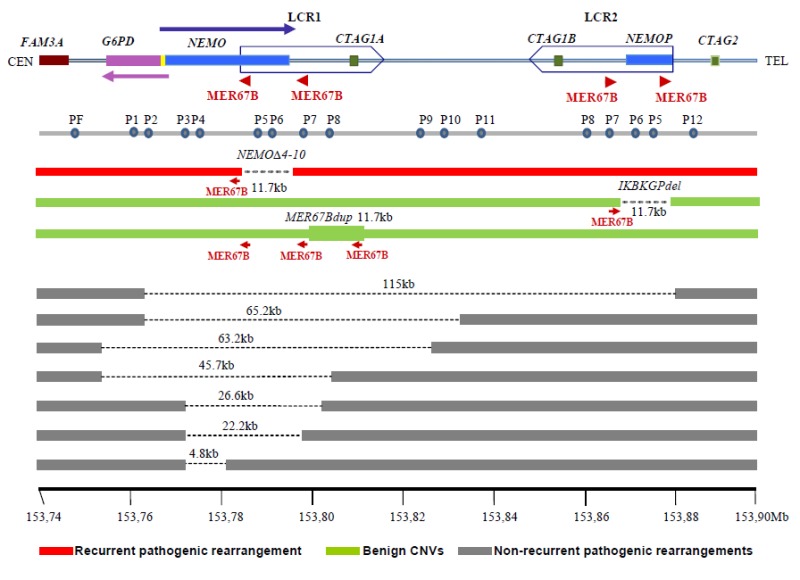
図6:IP座位にみられる反復性・非反復性の再構成
最上段の線は、互いに逆方向を向いたボックスで囲む形で、互いに逆方向のリピートが存在することを示したものである。このボックス内には、それぞれ2つずつMER67Bリピート(赤矢印)が存在する。欠失を明示するためにアッセイが行われたリアルタイムPCRのアンプリコンの位置を、PF、P1、P2等々で示した。詳細については、本文、ならびにFuscoら[2012]を参照されたい。
注:IKBKGは以前NEMO、IKBKGP1は以前NEMOPと呼ばれていたものである。
遺伝子構造
IKBKGは、異なるアイソフォームをコードする複数の転写バリアントを有している。転写バリアントNM_003639.3には10のエクソンが含まれている。遺伝子ならびにタンパク質の情報に関する詳細については、表Aを参照されたい。
遺伝子
IKBKGには、高度な類似性を有するIKBKGP1(以前はNEMOPと呼ばれていた)(図6)と呼ばれる偽遺伝子が存在する。これは、X染色体内の隣接領域に位置している。IKBKGP1は、IKBKGのエクソン3-10と高度に類似した配列を有する部分性偽遺伝子である。
IKBKGとIKBKGP1とは22kb隔たっており、両者の配列は互いに逆方向を向いている。
良性のバリアント
IP罹患者の10%-12%は、次の2つの良性のバリアントを有することがわかっている。
- IKBKGP1のエクソン4-10の11.7kbの欠失(図6に、上側の緑線で模式的に示した染色体)
- 正常なIKBKG遺伝子の下流に、エクソン4-10が複製されてできるMER67Bの重複(MER67Bdupと呼ばれる)(下側の緑線で模式的に示した染色体)
両バリアントとも、対照集団で稀にみられる正常バリアントであったという[Fuscoら2009]。こうしたデータから示唆されることは、IPの座位は、組換えを起こして反復性のバリアントを生じさせるような座位であって、これが子に11.7kbの病的欠失をdenovoに生じさせる「高リスク要因」になっているということである。
病的バリアント
IP罹患者に生じる反復性の病的バリアントは、IKBKGのエクソン4から10までの11.7kbの欠失である(図6の赤で模式的に示した染色体)(「分子レベルの病原」の項を参照)。図6に、IPを引き起こす非反復性の欠失のいくつかを併せて示した。
遺伝子内の小さな置換・欠失・重複は、IKBKG全体に散在している。エクソン10は極度のGCリッチ状態を示すが、ここには反復性の病的バリアントのクラスターが存在する[Fuscoら2008]。シトシンが7つ連続するエクソン10のモノヌクレオチド列に生じる遺伝子内欠失/重複も複数報告されている[Aradhyaら2001a,Fuscoら2008]。
タンパク質の活性低下(活性喪失まではいかない)をもたらすIKBKG(主としてエクソン10)のさらに小さな病的バリアントが報告されている[Zonanaら2000,Aradhyaら2001a,Döffingerら2001,Fuscoら2008]。こうした病的バリアントは、女性については疾患の程度が軽度となり、男性については乏汗性外胚葉異形成-免疫不全症(HED-ID)や免疫不全・大理石骨病・リンパ浮腫を伴う無汗性外胚葉異形成症(OLEDAID)として現れることで、生存の可能性が出てくる(「遺伝型-表現型相関」の項を参照)。詳細については、表Aを参照されたい。
表2:IKBKGの病的バリアント(一部)
| DNAヌクレオチドの変化 | 予想されるタンパク質の変化 | 参照配列 |
|---|---|---|
c.399-?_1260+?del(高頻度にみられる11.7kbの欠失) |
-- |
NG_009896.1 |
c.1167dupC |
p.Glu390ArgfsTer5 |
NM_003639.3 |
上記のバリアントは報告者の記載をそのまま載せたもので、GeneReviewsのスタッフが独自にバリアントの分類を検証したものではない。GeneReviewsは、HumanGenomeVariationSociety(varnomen.hgvs.org)の標準命名規則に準拠している。命名規則の説明については、QuickReferenceを参照のこと。
正常遺伝子産物
2.8kbのIKBKGcDNAは、419のアミノ酸から成るタンパク質、IKK-γ(核内因子κBessentialmodulatorタンパク質;NP_003630.1)をコードする。このタンパク質は酸性で、グルタミン酸残基、グルタミン残基を豊富に含み(それぞれ13%)、アミノ酸315-342部にロイシンジッパーモチーフをもつ[Yamaokaら1998]。IKKタンパク質にはα、β、γがあり、これらが1つの複合体を形成する[Rothwarfら1998,Liら1999,Hayden&Ghosh2008]。
IKK-γは、胚発生初期の始まりの時点から産生され、広汎に発現する[Aradhyaら2001b]。正常遺伝子産物は、複合体の形でNF-κBを活性化し、腫瘍壊死因子αにより誘導されるアポトーシスから保護する役割をはじめ、多くの機能を有する。
異常遺伝子産物
IKK-γの異常ないし欠損が生じることにより、正常なIKK複合体が形成されなくなる。そのため、IP罹患者の細胞は、NF-κBを正常な形で活性化する能力を失っている。NF-κBが活性化されることでアポトーシスからの保護が図られる。そのため、IP罹患者の細胞はアポトーシス促進性のシグナルに対する感受性が非常に高く、容易に細胞死を起こす[Smahiら2000]。
11.7kbの欠失が生じると、結果的にNF-κBを活性化する能力が失われることになり、アポトーシスに対する感受性が極度に高まることになる。IPは、男性については胚の段階で死亡し、女性についてはX染色体不活化に極端な偏りがみられるが、その原因はそういったところにある[Smahiら2000,Courtois&Smahi2006]。重度のIPをもたらすIKBKGの2つの病的バリアントに関して分子レベルの研究が行われたことで、多種多様な外的刺激に対して反応すべきNF-κBの活性化阻害が本疾患の根本原因であることが明らかとなった[Sebban-Beninら2007,Gautheronら2010]。
更新履歴:
- Gene Review著者: Angela Scheuerle, MD, FAAP, FACMG and Matilde Valeria Ursini, PhD.
日本語訳者: 河合美紀、大江瑞恵、倉橋浩樹(藤田保健衛生大学・総合医科学研究所・分子遺伝学研究部門)
Gene Review 最終更新日: 2010.10.28 日本語訳最終更新日:2014.9.23 Gene Reviews著者: Angela E Scheuerle, MD, FAAP, FACMG and Matilde Valeria Ursini, PhD. 日
本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(カレス記念病院ゲノム医療センター)
GeneReviews最終更新日: 2025.4.3. 日本語訳最終更新日: 2025.4.6.[in present]
| X POST |
![]()