22q11.2欠失症候群
(22q11.2 Deletion Syndrome)
[Synonyms:22q11.2DS]
Gene Reviews著者: Donna M McDonald-McGinn, MS, LCGC, Heather S Hain, PhD, LCGC, Beverly S Emanuel, PhD, and Elaine H Zackai, MD, FACMG
日本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日:2020.2.27. 日本語訳最終更新日:2024.4.7.
要約
疾患の特徴
22q11.2欠失症候群(22q11.2DS)罹患者の所見は、家系内においてさえ非常に大きなばらつきがみられる。22q11.2DSの主たる症候としては、先天性心疾患、中でも特に円錐動脈幹奇形(心室中隔欠損、Fallot四徴、大動脈弓離断、総動脈幹)、口蓋の異常(鼻咽腔閉鎖機能不全、粘膜下口蓋裂、口蓋垂裂、口蓋裂)、免疫不全、特徴的顔貌、学習障害が挙げられる。感音性、伝音性、混合性の難聴がみられることもある。喉頭気管食道奇形、消化器奇形、眼奇形、中枢神経系奇形、骨格奇形、尿路性器奇形がみられることもある。22q11.2DSにおいては、精神疾患や自己免疫疾患の頻度も高い。
診断・検査
22q11.2DSの診断は、染色体マイクロアレイ検査、もしくはその他のゲノム解析により、22q11.2のヘテロ接合性欠失を確認することで行われる。
臨床的マネジメント
症状に対する治療:
心奇形については心臓病専門医の推奨に従って治療を行う。口蓋奇形の修復術については、耳鼻科医の勧めに従って行う。摂食障害についてはスプーンの持っていき方の工夫で対応し、胃食道逆流症や消化管運動異常については標準治療を行う。免疫不全については、感染に関する積極的な治療が必要で、稀には抗生剤の予防投与、免疫グロブリン静注療法、胸腺移植が必要になるようなこともある。免疫系の機能が正常であることが確認されるまでは、放射線照射血液製剤の使用が推奨される。自己免疫疾患については、個々の免疫医ごとの推奨治療を行う。カルシウム補給については、もし長期投与が必要であれば、腎結石のリスクを考慮し、内分泌内科医や腎臓専門医への紹介を行う。成長ホルモン分泌不全に対しては標準治療を行う。眼奇形についても標準治療を行う。難聴に対しては補聴器の使用が有用である。作業療法、理学療法に加え、1歳までに手話を交えて開始する言語治療、教育療法、行動療法を行う。精神疾患に対しては、必要に応じた支援や治療を行う。頸椎奇形に対しては、整形外科医の推奨に基づいて活動制限を行う。腎奇形については、腎臓専門医の勧めに従って手術や治療を行う。シーラントを含む通常の歯科治療を行う。
定期的追跡評価 :
言語の表出開始以降は、スピーチの鼻音化に関する評価を行う。抗体陽転の評価のため、抗体検査を行う。小児期は、生ワクチン投与前に免疫の状態を再評価する。年に1度、全血算、白血球分画の検査を行う。乳児期には3ないし6ヵ月ごと、小児期には5年ごと、その後は1,2年ごと、また術前術後、妊娠中は定期的に、それぞれ血清カルシウム濃度の測定を行う。年に1度、甲状腺刺激ホルモン(TSH)と遊離T4の検査を行う。1歳から3歳までの間、ないし必要に応じた形で眼科的評価を行う。乳児期、就学前期、ならびに学齢期に聴覚検査を行う。年に1度、発達評価を行う。脊柱側彎に関して年に1度の追跡評価を行う。6ヵ月ごとに歯科検診を行う。
避けるべき薬剤/環境 :
リンパ球異常を示す乳児に対しては生ワクチン(経口ポリオワクチン、MMR3種混合ワクチンなど)の投与を避ける。炭酸飲料、飲酒は低カルシウム血症を悪化させる可能性がある。カフェイン摂取により不安をきたしたり、不安を増悪させたりする可能性がある。
遺伝カウンセリング
22q11.2DSは、常染色体顕性の隣接遺伝子欠失症候群である。3.0(2.54)Mbの欠失に起因して生じる22q11.2DSについては、90%以上の例がdenovoの欠失例で、約10%がヘテロ接合体の親からの継承例である。22q11.2の入れ子型欠失(nesteddeletion)を有する22q11.2DS罹患者については、その60%が罹患者の親からの継承例である。罹患者の子が22q11.2の欠失を継承する可能性は50%である。家系内に存在する22q11.2の欠失が確定している場合は、FISH法やMLPA法を用いた出生前検査、あるいは高リスクの妊娠に備えたアレイ検査や着床前遺伝学的検査を行うことが可能である。
GeneReviewの視点
| 22q11.2欠失症候群:この中に含まれる表現型 |
|---|
|
同義語、ならびに過去に用いられた呼び名については、「疾患名について」の項を参照のこと。
診断
本疾患を示唆する所見
以下のような臨床所見を示す患者については、22q11.2欠失症候群(22q11.2DS)を疑う必要がある。
臨床症候
- 先天性心疾患(罹患者の64%にみられる)、特に円錐動脈幹奇形(例えば心室中隔欠損、Fallot四徴、大動脈弓離断、総動脈幹)
- 鼻咽腔閉鎖機能不全、粘膜下口蓋裂、口蓋垂裂、口蓋裂、開鼻声、嚥下障害などの口蓋の異常(67%)
- 血管輪、喉頭横隔膜症、喉頭気管軟化症、声門下狭窄などの喉頭気管食道異常
- 消化器の構造的異常(例えば、前方肛門や鎖肛、食道閉鎖、空腸閉鎖、腸回転異常、Hirschsprung病)を伴うことのある便秘、副脾、横隔膜ヘルニア、臍ヘルニア、鼠径ヘルニアなどの消化管奇形
- 免疫不全(77%)(例えば高頻度の感染、胸腺形成不全)
- 自己免疫疾患(例えば若年性関節リウマチ、バセドウ病、白斑)
- 網膜血管の蛇行、眼瞼下垂、後部胎生環、強膜化角膜、コロボーマ、白内障、無眼球、斜視等の眼科的所見
- その他の頭蓋顔面所見(例えば、フード状眼瞼、耳奇形、突出した鼻梁、球状の鼻、小下顎症、啼泣時の顔面非対称、頭蓋縫合早期癒合症)
- 難聴(感音性、伝音性、混合性)
- 乳児期の筋緊張低下、小頭症、多小脳回、てんかん発作(特発性ないし低カルシウム血症関連のもの)
- 発達障害ないし学習障害(70%-90%)、特に非言語性学習障害
- 自閉症スペクトラム障害(小児の20%)、統合失調症(成人の25%)、注意欠如障害、不安、固執、社会的相互関係構築困難などの精神疾患
- Parkinson病の早発
- 後頭骨-頸椎奇形、脊柱側彎、肋骨・椎骨奇形、内反足、多指趾症などの骨格奇形
- 腎奇形(16%)(例えば水腎症、腎無発生、多嚢胞性腎/異形成腎)、停留精巣、尿道下裂などの尿路性器奇形
検査所見
- 副甲状腺機能低下症と低カルシウム血症(50%)
- 成長ホルモン分泌不全症
- 甲状腺機能低下症
- 血球減少症(溶血性貧血、好中球減少症、血小板減少症)
診断の確定
発端者における22q11.2DSの診断は、22q11.2のヘテロ接合性欠失を確認することで確定する(表1参照)。22q11.2DSを有する大多数の罹患者(85%近く)は、染色体マイクロアレイで、参照ゲノム(NCBIBuildGRCh37/hg19)における隣接低コピー反復配列A-D部の、TBX1を含むおおむねg.18,912,231-21,465,672の位置における2.54Mbのヘテロ接合性欠失を示す(図1)。
注:歴史的には、反復欠失は3Mbのサイズの欠失とされてきた[Guoら2016]。
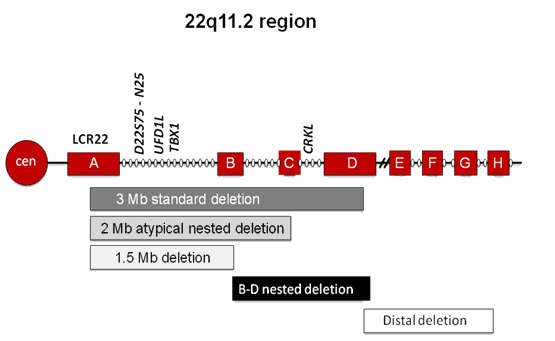
図1
罹患者の大多数(85%)は、40の遺伝子を含む2.54Mbの欠失を示す。ただ、「入れ子型(nested)」欠失とも呼ばれるもっと小さな非定型欠失を示すタイプもある。McDonald-McGinn&Zackai[2008]より、WileyandSons社の許可を得て転載。
注:22q11.2DS罹患者の約5%は、低コピー反復配列A-B部の1.5Mbのヘテロ接合性欠失を示す。また、約2%は低コピー反復配列A-C部の欠失、約5%はさらに小さな低コピー反復配列B-D部ないしC-D部のヘテロ接合性欠失を示す。
この欠失をISCNの表記法で表すと、以下のようになる。
- 2.54Mbdel:seq[GRCh37]del(22)(q11.2)chr22:18,912,231-21,465,672
- 1.5Mbdel:seq[GRCh37]del(22)(q11.2)chr22:20,731,986-21,465,672
ゲノム検査の手法
配列のコピー数を判定するゲノム検査の手法としては、染色体マイクロアレイ(CMA)や標的型の欠失解析がある。
注:22q11.2の反復欠失は、通常の染色体Gバンド分染法その他の従来型細胞遺伝学的分染法では同定することができない。
- 染色体マイクロアレイ
発端者の有する反復性の欠失は、オリゴヌクレオチドやSNPのアレイを用いたCMAで検出可能である。欠失のサイズを測る能力は、使用するマイクロアレイの種類、ならびに22q11.2領域のプローブの密度によって決まる。
注:(1)22q11.2の反復性の欠失を有する罹患者の大多数は、発達遅延、知的障害、自閉症スペクトラム障害の評価の流れで行うCMAにより同定がなされる。(2)2004年以前の段階では、多くのCMAプラットフォームはこの領域をカバーしていなかったため、この領域の欠失を同定することができなかった。22q11.2の欠失を調べる初期のアレイは、バクテリア人工染色体比較ゲノムハイブリダイゼーション(BACCGH)アレイで、その解像度は25kb程度、あるいはそれ以下であった[Mantripragadaら2004]。
- 標的型の欠失解析
22q11.2の反復性の欠失が判明している発端者の血族を調べるときは、FISH法、定量的PCR(qPCR)、MLPA法その他の標的型の定量的手法が使用可能である。
注:(1)22q11.2領域を標的とするCMAで反復性の欠失が同定されなかった被検者に対して、標的型の欠失解析を行うのは適切でない。(2)標的型の手法を用いることで、欠失の大きさを定常的に測ることが可能である。特に、MLPA法はLCRごとにサイズの異なる欠失を同定する上で有用である。
表1:22q11.2欠失症候群に用いられるゲノム検査
| 欠失1 | 手法 | 感度 | |
|---|---|---|---|
| 発端者 | リスクを有する血族 | ||
22q11.2領域の2.54Mbのヘテロ接合性欠失
Chr22:18,912,231-21,465,672del2
|
CMA3 |
100% |
100% |
標的型の欠失解析4 |
95%-100%近く5 |
100%6 |
|
- この領域の欠失、ならびにここに含まれる注目すべき遺伝子の詳細については、「分子遺伝学」の項を参照。
- ClinicalGenomeResource(ClinGen)プロジェクト(旧InternationalStandardsforCytogenomicArrays(ISCA)コンソーシアム)の表記によるゲノム変異の標準的なISCN表記とその解釈。ここに示したゲノム座標の数字は、ClinGenの方式で表した22q11.2の最小の反復性欠失である。欠失の座標上の数字は、検査機関のアレイデザインにより若干異なることがある。切断点の近傍には分節重複が存在するため、予想される欠失のサイズと座標から算出される欠失のサイズとの間に相違が生じることがあることに注意が必要。この領域内におけるこれより有意に大きい、もしくは有意に小さい欠失に伴って生じる表現型は、22q11.2反復性欠失とは明確に異なるものであることが考えられる(「遺伝学的に関連のある疾患」の項を参照)。
- オリゴヌクレオチドあるいはSNPのアレイを用いた染色体マイクアレイ解析(CMA)。現在、臨床で使用されているCMAは、22q11.2領域を対象の1つにしたデザインとなっている。
- 標的型欠失解析法としては、FISH法、定量的PCR法(qPCR)、MLPA法、ならびにその他の標的型定量的手法などがある。
- 現在、商業ベースに乗っているFISHプローブであるN25やTUPLEは、TBX1ともども、低コピー数反復配列(LCR)のA-B間にあるため、LCRA-Bを含まない非定型の欠失については、市販のFISHプローブでは検出することができない(図1)。
- すでに22q11.2の反復性の欠失が判明している発端者の、リスクを有する血族に対しては、標的型のFISH、MLPA、その他の定量的解析手法を用いることが考えられる。
リスクを有する血縁者の検査
リスクを有する発端者の血族における22q11.2の反復性の欠失の確認には、FISH法、定量的PCR、MLPA法その他の、定量的な標的型欠失解析の手法が用いられる。再発率を把握する上で、親のサンプルを検査することは重要である。
臨床的特徴
臨床像
このセクションでは、22q11.2欠失症候群(22q11.2DS)を有する罹患者についてこれまでに報告されてきた所見を要約して記述する。
心臓
22q11.2DSの1,421人の記録を総括した報告によると、64%に先天性心疾患(CHD)がみられたという[Campbellら2018]。最も高頻度にみられたのは、流出路の円錐動脈幹奇形である(表2参照)。心エコーで確認される異常としては、心室中隔欠損が最も多い。CHDは、22q11.2DSを有する子どもの主たる死因(全死亡例の87%に及ぶ可能性)になっている[McDonald-McGinnら2015]。加えて、22q11.2DSの罹患者は成人についても若年での死亡がみられ、その原因は、CHDを伴わない罹患者においてさえ、突然死ならびに心不全が最大要因となっている[Bassettら2009]。罹患者の一部に大動脈基部拡張を示すサブセットが存在することがわかっているが[Johnら2009]が、大動脈基部拡張の自然経過についてはよくわかっていない。
表2:22q11.2欠失症候群罹患者の心所見
| 口蓋の所見 | 罹患者に占める割合 |
|---|---|
| 口蓋裂(CP) | 28.5% |
| ・口唇口蓋裂(CLP)1 | 0.6% |
| ・通常の口蓋裂 | 4.4% |
| ・粘膜下口蓋裂(SMCP)2 | 22.8% |
| 口唇裂単独 | 0.2% |
| 鼻咽腔閉鎖機能不全 | 55.2% |
| ・CP+VPI | 3.5% |
| ・SMCP+VPI | 18.4% |
| ・CPを伴わないVPI | 33.3% |
| 要追跡/低年齢のため評価不能3 | 18.7% |
| 異常なし | 26.6% |
- 片側性、両側性の両方を含む。
- 内訳は、典型例(5.1%)、潜在型(2.4%)、口蓋垂裂(13.6%)。
- 明らかな異常はみられないものの、子どもが幼すぎて十分な音声サンプルを採取できない例、ならびに協力が得られない例。
| 心所見 | 罹患者に占める割合 |
|---|---|
| 心室中隔欠損 | 23% |
| Fallot四徴(TOF)1 | 18% |
| 大動脈弓奇形2 | 14% |
| 大動脈弓離断(IAA) | 11% |
| 心房中隔欠損 | 10% |
| 肺動脈閉鎖 | 6% |
| 総動脈幹(TA) | 4% |
| 動脈管開存 | 6% |
| 大動脈二尖弁 | 3% |
| 肺動脈弁狭窄 | 2% |
| その他 | 1% |
摂食
約36%の患児に重大な摂食障害がみられ、嚥下障害のため経鼻胃管栄養や胃瘻造設術が必要になるような状況がしばしばみられる。摂食障害は、心疾患や口蓋の異常を伴わない罹患者においてもみられることが報告されている。そうした患児をさらに調べてみると、咽頭-鼻腔逆流傾向、輪状咽頭筋隆起、輪状咽頭部の異常閉鎖、憩室形成がしばしば明らかになる。そうしたことから、摂食障害の背景にあるものは、多くの患児の場合、第3・第4咽頭嚢由来の咽頭食道領域の運動障害であるように思われる。そして、誤嚥が呼吸困難や反復性肺感染症、反応性気道疾患の原因になっているものと考えられる[Eicherら2000]。
罹患者の大多数にみられる慢性所見に便秘がある。さらに、構造的異常としては、鎖肛、腸回転異常、腸無回転、先天性横隔膜ヘルニア、食道閉鎖、気管食道瘻、Hirschsprung病、血管輪から続発性に生じる摂食障害などが報告されており、これらが摂食嚥下障害の遠因になり、症例によっては便秘の原因にもなっている可能性がある[Digilioら1999,Kilicら2003,Jyonouchiら2009,Campbellら2018]。
免疫機能
免疫機能不全は、胸腺形成不全とそれに続くT細胞産生障害の結果として生じる。22q11.2DSの新生児は、胸腺系統の細胞が有意に少ない。これまでの研究で、罹患者の67%はT細胞の産生障害、19%はT細胞の機能障害を示すとされている[Smithら1998,Sullivanら1999,Sullivan2019]。22q11.2DS罹患者1,421人について行った研究では、50%にT細胞数の異常がみられた[Campbellら2018]。T細胞の産生は経時的に改善し、最重度のT細胞産生障害を示す罹患児でも、満1歳までにほぼ改善が得られていた[Sullivanら1999]。T細胞数の軽度減少を示す罹患児については、その多くが病原体に対する正常な防御能を有していた[Sullivanら1999]。
TBX1欠失に伴い、CD4陽性リンパ球減少症が生じる。1歳前後の罹患児52人を対象とした研究で、TBX1の欠失を示さない罹患児(入れ子型欠失もしくは遠位の欠失)に比べ、TBX1の欠失を有する罹患児(A-B,A-C,A-Dといった近位の欠失)のほうが、CD3細胞数やCD4細胞数が有意に少なかった[Crowleyら2018]。
液性免疫の異常が罹患者の17%で認められている[Campbellら2018]。IgA欠損症が13%で報告されており、中でも若年性関節リウマチ(JRA)をはじめとする自己免疫疾患を有する罹患者で特に多くみられる[Sullivan2019]。満1歳未満の段階でみられる低γグロブリン血症は、その後ふつう改善し、時には5歳以降に高γグロブリン血症となる例もみられる。罹患者の大多数については、抗体の機能やアビディティは正常であるが、中には抗体の機能に異常を有する例もある。反復性の洞肺感染症を示す罹患者には、しばしば免疫グロブリンの異常、特に肺炎球菌莢膜ポリサッカライドワクチンに対する抗体の反応障害がみられる[Genneryら2002,Sullivan2019]。
特に先天性心疾患を有する罹患者については、口蓋の機能障害、胃食道逆流症、誤嚥性肺炎などがすべて反復性の感染症につながる。さらに、嚥下障害により栄養不良が引き起こされ、それによってさらに細胞性免疫が低下する。このような形で、年長の子どもや成人でも感染は継続し、25%-33%に反復性の副鼻腔炎ないし中耳炎、4%-7%に反復性の下気道感染症がみられる[Jawadら2001]。ただそれでも、学齢期の子どもでは、免疫不全に関して何らかの積極的な処置を要するといった例はほとんどみられない[Sullivan2019]。
自己免疫疾患
T細胞リンパ球減少症に関連して生じる続発性のものとしては、アトピーや自己免疫疾患のリスクの上昇がある。JRAは、一般集団に対して、22q11.2DSの子どもでは20倍の発生頻度を示す。JRAの発症年齢は17ヵ月から5歳の間である。JRAはしばしば多関節にわたり、対処が容易でないことも多い。JRAの発症しやすさに関して、ヒト白血球抗原(HLA)が関与しているとの報告がある[Sullivanら1997]。22q11.2DSに関連して生じるその他の自己免疫疾患としては、特発性血小板減少性紫斑病(ITP)、甲状腺機能亢進症(Graves病)、甲状腺機能低下症、皮膚白斑、溶血性貧血、自己免疫性好中球減少症、再生不良性貧血、セリアック病などがある。ITPは、22q11.2DSでは一般集団の200倍の発生頻度を示す[Jawadら2001,Kawameら2001,Sullivan2019]。
副甲状腺機能
副甲状腺機能低下症と、それに続く低カルシウム血症が、22q11.2DS罹患者の17%-60%にみられ、その程度は、一般的には新生児期に最も顕著に現れる。カルシウムホメオスタシスの問題は、年齢とともに正常化していくことが多いものの、小児期後期や成人期においても、疾病罹患時、思春期、妊娠時などに再度現れることが報告されている。
Cheungら[2014]は、22q11.2DSの成人の80%は、生涯のある時期に低カルシウム血症を経験するとした上で、低カルシウム血症に至る要因として、副甲状腺機能低下症、甲状腺機能低下症、低マグネシウム血症が関与すると述べている。
成長
22q11.2DSの成人罹患者の大半は正常の身長を有する。しかし、1歳から15歳までの95人の子どもにおいて、その41%が5パーセンタイル未満の身長を示したという報告があり、そのうちの4人は5パーセンタイルをはるかに下回り、4人ともIGF1、IGFBP3が低値を示した。成長ホルモン分泌不全が3人、MRI上での下垂体の萎縮が3人にみられた。そして、2人がヒト成長ホルモンの投与に良好な反応を示した[Weinzimerら1998]。22q11.2DS罹患者の成長曲線が公表されている[Habelら2012]。22q11.2DSの成人の35%に肥満がみられたとの報告がある[Bassettら2005]。
眼
90人を対象とした眼の異常に関する前向き研究で、フード状眼瞼(20%)、眼瞼下垂(4%)、睫毛重生(マイボーム腺開口部から異常な形で睫毛が成長する状態)(2%)が報告されている。その他の所見としては、後部胎生環(49%)、角膜神経の拡大(3%)、虹彩窩の拡大(2%)、網膜血管の蛇行(34%)、傾斜視神経乳頭(1%)がみられた。斜視の出現は18%、弱視は4%にみられた。また少数の罹患者に白内障とコロボーマがみられた[Forbesら2007]。後部胎生環がみられたのは対照群では12%-32%であったが、22q11.2DS罹患者では、Alagille症候群(89%)[Krantzら1997]にほぼ匹敵する高値を示した。乱視、近視、遠視については、一般集団との差はみられなかった。ごく少数の患者で、無眼球がみられている[著者の確認したものであるが、未公表]。
他の頭蓋顔面所見
口蓋と眼の奇形以外の頭蓋顔面所見としては、耳奇形、鼻奇形、啼泣時の顔面非対称、小下顎症、頭蓋縫合早期癒合症などがある[McDonald-McGinnら2015]。耳奇形としては、過剰に折れ込んだ耳輪ないし四角い耳輪、カップ耳・小耳・立ち耳、耳前部の瘻孔ないし副耳、外耳道狭窄などがある。感音性難聴・伝音性難聴の両方の報告がある。突出した鼻梁、球状の鼻、鼻翼低形成、鼻尖の陥凹/鼻尖裂が多くみられる[Campbellら2018]。血管輪・喉頭軟化症・喉頭横隔膜症に起因する喘鳴、喉頭閉鎖、声門下狭窄もみられることがある。
顔面所見はばらつきが大きく、特にアフリカ系の起源をもつ罹患者については、特徴的な所見がみられないことも多い[Kruszkaら2017]。
中枢神経系
22q11.2DS罹患者の大多数は、乳児期の筋緊張低下の既往を有する[Swillen&McDonald-McGinn2015]。啼泣時の顔の非対称については、罹患者の8%-14%にみられるとの研究報告がある[McDonald-McGinnら1997,Campbellら2018]。小頭症については、罹患者の24%-50%にみられるとされている[McDonald-McGinn&Sullivan2011,Campbellら2018]。てんかん発作は、低カルシウム血症に付随して生じることが最も多い。ただ、22q11.2DS患者の約15%は、非誘発性発作を示す[Fungら2015]。
22q11.2DSの24症例を調べたMRI/MRAの研究で、透明中隔腔ないしベルガ腔の遺残(8/24)、多小脳回あるいは皮質形成異常(4/24)、小脳形成不全(1/24)など、半数を超える例(13/24)に大きな異常所見がみられた[Bohmら2017]。fMRIにて、同年齢・同性の対照と比較して、脳の後部の体積が有意に少なく、前頭葉に対して左後頭部や左頭頂部により顕著な白質の減少がみられた[Beardenら2004,Bishら2004,Katesら2004]。拡散テンソル画像を用いて行われた大規模多施設研究によると、22q11.2DS罹患者では、特に皮質-皮質間経路において、平均拡散、軸拡散、放射拡散に広範囲の減少がみられている[Villalon-Reinaら2019]。
心理社会的発達ならびに認知機能
一般に、22q11.2DSの幼い子どもの運動発達指標には遅れがみられ、歩行開始は18ヵ月である。また、発語にも遅れがみられ、多くの罹患児は2-3歳の段階では発語未開始である。
55人の幼児に対してBayley発達検査を行った報告によると、精神発達は正常が22%、軽度遅延が20%、明らかな遅延が58%であった。精神発達指数の平均は、明らかな遅延にあたる67±15で、精神運動発達指数の平均は61±13であった。スピーチや言語の遅れは、評価したすべての子どもにみられている。同じ研究で、WPPSI-R(訳注:ウェクスラー幼児知能検査の改訂版)を用いて評価を行った24人の就学前の子どもの群については、全検査IQが78±12、平均動作性IQが78±12、平均言語性IQが81±13であった。言語全体では、20%が平均レベル、46%が軽度遅延、34%が明らかな遅延であったが、点数は、表出言語より受容言語のほうが高かった[Gerdesら2001]。
80人の学童期の子どもに対して、ウェクスラー知能検査で評価を行った研究では、平均IQは76.8±13.0で、39%が平均レベル、31%が平均低値のレベル、31%が境界レベルであった[Niarchouら2014]。
それ以降の年齢の22q11.2DS患者は、一般に、複数のドメインにわたる非定型的な神経心理プロファイルを示す。その中で最も特徴的なのは、動作性IQに比して有意に高い言語性IQを示すことである。Mossら[1995]は、80人の学童期の子どもの66%に、言語性IQと動作性IQの平均値の乖離があったと報告しており、障害があったのは常に非言語性IQのほうであったと述べている。この現象は、一般集団では稀とされている[Swillenら2018]。22q11.2DS罹患者の多くについては、全検査IQスコア単独ではその知能の状態を正確に表現することができないため、言語性IQと動作性IQを、それぞれ別個に評価する必要がある。さらに、罹患者は機械的繰り返しによる言語習得と言語記憶、読む力(訳注:日本語に置き換えるのが難しいが、原文は「readingdecoding」、すなわち「書かれた綴りを見て、それをどう発音するべきかを正しく判断する能力」のこと)、綴り方の力といった分野に相対的な強みを示す。一方で、非言語的情報処理、視覚的空間認識能力、複雑性の高い言語の記憶力、注意力、ワーキングメモリ、視覚-空間メモリ、計算といった分野に弱みをもつ。視覚メモリより言語メモリの能力に強みがあり、計算より読む力のほうに強みがあるという事実からも、非言語性学習障害の存在が裏打ちされる。そのため、こうした背景に特化した認知矯正療法、行動管理、親へのカウンセリングといったことが求められる。
精神疾患
22q11.2DS罹患者の中には、一方で脱抑制・衝動性、他方で内気・引きこもりといった行動・性格を示す例がみられる。注意欠如、不安、固執、社会的交流の難しさといったものも多くみられる。自閉症/自閉症スペクトラム障害が罹患者の20%で報告されている[Swillen&McDonald-McGinn2015]。
成人罹患者の60%に精神疾患がみられるとされている。特筆すべきは、統合失調症が罹患者の約25%にみられることである。ただ、それだけでなく、不安障害や抑鬱障害もかなり多くみられる[Bassetら2011]。行動様式の違いは幼少期に始まる。そのため、22q11.2DSの子どもについては、10歳までの段階で精神医学的スクリーニングを行うことで、早期治療の可能性が生まれるのではないかと思われる。
Parkinson病の早発
22q11.2DSにはParkinson病の早発リスクがあるとされており、5.9%という発症率が報告されている[Fungら2015]。その他の神経変性疾患については、現在のところほとんど報告されていない。
筋骨格系
22q11.2DSの筋骨格系の症候については、1つの確かな実証が存在する。それは、後頭骨・頸椎部の画像検査を行った罹患者(10研究408人)の90.5%-100%に1つ以上の後頭骨-頸椎異常がみられたということである[Homansら2018]。よくみられたものは、頭蓋の扁平化、第1頸椎の形態異常ないしアーチの閉鎖不全、第2頸椎の椎弓板や後方部の反り上がりを伴う歯突起の形態異常であった。前屈・後屈のX線写真でしばしば報告されている奇形は、分節運動の過大(56%)である。脊柱側彎が14の論文(2,264症例)で報告されており、出現頻度は0.6%-60%とされている。肋骨奇形は2つの論文で報告されており、出現頻度は2%-19%となっている。椎骨の異常は1.1%-11%と報告されている。
四肢に現れる症候で最もよくみられるのは内反足で、これは15の論文(2,115症例)で報告されており、出現頻度は1.1%-13.3%となっている。膝蓋骨脱臼は3つの論文で報告されており、出現頻度は10%-20%となっているが、これはエビデンスとしては弱い。報告されているその他の異常としては、多指趾症、肩の変形、足趾の重なりがある。
尿路性器
22q11.2DSの16%に腎奇形がみられたという報告がある。最も頻度の高い奇形は、片側性水腎症、腎無発生、多嚢胞性異形成腎であった[Campbellら2018]。上部尿路では水腎症が最も多くみられた所見であるものの、大多数(63%)において単独所見としての上部尿路の拡大を認めた。尿路性器異常と診断されるのは、女児より男児のほうが有意に多い(0.5%対8%)傾向がみられた。男児では、4%に停留精巣、4%に尿道下裂がみられた。女児にみられたその他の異常としては、膣無発生が2例、子宮無発生が1例であった。その他に報告されているものとしては、臍ヘルニア、鼠径ヘルニア、尿道索、包茎、停留精巣(訳注:すぐ前にも「停留精巣」の記述があり、記述が重なっている。前のものは「cryptorchidism」、後のものは「undescendedtestis」で、何らかの使い分けがあるのかもしれないが、ここではどちらも「停留精巣」と訳した)などがある。
その他
22q11.2DS罹患者において報告されているその他の所見:
- 肺分葉異常[McDonald-McGinnら1995]
- 歯の齲蝕
- 肝芽腫、腎細胞癌、甲状腺癌、黒色腫、白血病、Wilms腫瘍、神経芽腫などの悪性腫瘍[Campbellら2018,Lambertら2018,]。全体としての発生頻度は、約6%である。
- Bernard-Soulier症候群(GP1BBの病的バリアントに起因)やCEDNIK症候群(SNAP29の病的バリアントに起因)などの常染色体潜性遺伝疾患(22q11.2DSの罹患者において、残るもう一方のアレルに病的バリアントが生じた場合に発生することが報告されている)
遺伝型-表現型相関
CD4陽性リンパ球減少症と欠失の切断点との相関が報告されている。22q11.2の入れ子型欠失あるいは遠位の欠失(B-D、C-D、D-E、D-F)を有する罹患者に比べ、22q11.2の近位の欠失(A-B、A-C、A-D)を有する罹患者は、CD3、CD4細胞数が有意に少なかった[Crowleyら2018]。
浸透率
22q11.2DS罹患者の大多数については、浸透率は100%であるが、表現度に非常に大きな幅がみられる。入れ子型欠失はしばしば家族性に出現するが、このタイプについては浸透率が100%ではなく、表現度も低いことが多い。
疾患名について
現在22q11.2DSは、以前DiGeorge症候群(DGS)、軟口蓋心臓顔面症候群(VCFS)、円錐動脈幹異常顔貌症候群(CTAF)、ならびに一部の常染色体顕性遺伝型OpitzG/BBB症候群、Cayler心臓顔面症候群(啼泣時の顔面非対称)と呼ばれていた表現型をすべて包含するものと捉えられている[McDonald-McGinnら2015]。上記のさまざまな臨床病名は、確認バイアスの結果として生じたものである。
「DiGeorge症候群」という病名は、現在では、22q11.2の欠失を有しないにもかかわらず22q11.2DSの臨床症状を呈する例について用いるものとされている。
「常染色体顕性遺伝型OpitzG/BBB症候群」という病名は、眼間開離、食道奇形、尿道下裂を有する例について用いられる。
頻度
22q11.2DSは、染色体微小欠失症候群の中で最も高頻度にみられるものである。スウェーデンで行われた人口ベースの研究で、平均年間発生数は100,000生産児あたり14.1人とされている[Oskarsdóttirら2004,Oskarsdóttirら2005a,Oskarsdóttirら2005b]。アメリカ合衆国で行われた人口ベースの研究では、白人・黒人・アジア人については6,000人に1人、ヒスパニック系については3,800人に1人という数字が出ている[Buttoら2003]。
遺伝学的に関連のある疾患(同一アレル疾患)
22q11.2症候群には、このGeneReviewで述べたもの以外の表現型はみられない。
22q11.2DSの臨床所見を示す罹患者のごく少数(1%未満)は、22番染色体と別の染色体との間の転座など、22q11.2領域を含む染色体再構成の例である。
鑑別診断
22q11.2欠失症候群(22q11.2DS)で現れるすべての臨床所見は、その1つの所見以外には特段の異常がみられない単発性奇形の形で現れることがある。このセクションでは、22q11.2DS類似の臨床的表現型を示す遺伝性疾患ないし催奇形因子への曝露について述べる。
単一遺伝子疾患
表4:22q11.2欠失症候群との鑑別診断の対象となる遺伝子
| 遺伝子 | 疾患名 | 遺伝形式 | 22q11.2欠失症候群と重なる所見 |
|---|---|---|---|
| CHD7 | CHARGE症候群 | AD | 先天性心疾患,口蓋異常,コロボーマ,後鼻孔閉鎖,成長障害,耳奇形ないし難聴,発達遅延,顔面麻痺,尿路性器奇形,免疫不全 |
| DHCR7 | Smith-Lemli-Opitz症候群 | AR | 多指趾,口蓋裂 |
| JAG1 NOTCH2 |
Alagille症候群 | AD | 蝶形椎,先天性心疾患,後部胎生環 |
| TBX11 | Fallot四徴 (OMIM187500) |
AD | 先天性心疾患,耳前瘻孔 |
AD=常染色体顕性;AR=常染色体潜性
- TBX1は、22q11.2のA-B領域内に座位する遺伝子であるが、22q11.2欠失症候群の臨床所見(主として先天性心疾患)を有するものの、22q11.2の欠失は示さない罹患者の中に、このTBX1の病的バリアントをもつ例があることが知られている[Gongら2001,Yagiら2003]。
染色体異常
10p13-p14欠失
22q11.2DSと重なる所見として、心奇形、免疫不全、副甲状腺機能低下症、口蓋裂、発達遅滞、小頭症、停留精巣などがある[Lichtnerら2000]。
11q23-ter欠失(Jacobsen症候群)(OMIM147791)
22q11.2DSと重なる所見として、小頭症、小下顎症、耳介低位、眼症状、心奇形、尿道下裂、停留精巣、免疫不全などがある。
その他
病因遺伝子不明の疾患
- VACTERL連合(先天性心疾患、脊椎奇形、腎奇形、四肢奇形がみられるときに鑑別対象となる)。VATER連合が1つの除外診断であるが、現在、病因は確定していない(OMIM192350)。
- 眼-耳-脊椎症候群(OAVS)(Goldenhar症候群)(耳奇形、脊椎奇形、心疾患、腎奇形がみられるときに鑑別対象となる)(OMIM141400)。
催奇形因子への曝露
22q11.2DS類似の表現型は、母体の糖尿病、ならびに母体のレチノイン酸曝露に伴って現れることがある。
臨床的マネジメント
最初の診断に続いて行う評価
すでに、22q11.2欠失症候群(22q11.2DS)罹患者の評価と治療に関するガイドラインが公表されている。Bassettら[2011](全文)ならびにFungら[2015]を参照されたい。
22q11.2DSと診断された罹患者については、疾患の範囲ならびに治療上のニーズを把握するため、診断に至る過程ですでに実施済でなければ、表5にまとめた評価を行うことが推奨される。
表5:22q11.2欠失症候群罹患者の最初の診断後に行うことが推奨される評価
| 系/懸念事項 | 評価 | コメント |
|---|---|---|
| 心臓 | 胸部X線写真・心電図・心エコーを含む心臓病専門医による評価 | 血管輪が疑われるときは、胸部MRIが必要なこともある。 |
| 耳鼻咽喉系 |
|
|
| 消化器系 | 摂食の問題の評価(例えば、胃食道逆流症や吸啜・嚥下障害の有無、離乳食への移行状況、食感を残した食材の添加状況、嘔吐の有無)、ならびに便秘に関する評価 | 必要に応じ、解剖学的異常について調べる。 |
| 免疫系 | 白血球百分率を含む全血算(CBC) |
|
| 血液系 | 内出血/出血多発の既往のある罹患者については、血液内科医による評価 | 外科手術を行う前に、血小板の量・機能の評価を考慮する。 |
| 内分泌系 |
|
|
| TSHならびに遊離T4 | 甲状腺機能低下症/亢進症の評価のため | |
| 成長評価 | 身長が2パーセンタイル未満の場合、成長ホルモン分泌不全の評価のため、内分泌内科医に紹介する。 | |
| 眼 | 眼科的評価 | 診断時に行う。 |
| 聴覚 | 聴覚評価 | 診断時に行う。 |
| 神経系 | 神経学的評価 | てんかんが疑われる場合に行う。 |
| 発達 | 言語評価 |
|
| 精神系 | 臨床心理士ないし精神科医による評価 |
|
| 骨格系 | 胸部X線写真 | 胸椎の異常を評価するため |
| 頸椎のX線写真(6方向:屈曲位・伸展位・AP・側面・開口位・頭蓋底) | 4歳(頸椎の骨化年齢)を過ぎた全患者、ならびに外科手術や運動(タンブリング等)で頸の過伸展を伴う際に前もって行う。 | |
| 腎臓 | 腎超音波検査 | |
| その他 | 臨床遺伝医や遺伝カウンセラーへの紹介 |
症候に対する治療
表6:22q11.2欠失症候群罹患者の症候に対する治療
| 症候/懸念事項 | 治療 | 考慮すべき事項/その他 |
|---|---|---|
| 心奇形 | 心臓病専門医の勧めに従った手術ないし治療 | 生涯のうちに複数の手術が必要になることがある。 |
| 口蓋奇形 | 耳鼻科医の勧めに従った手術 | 手術に先立って心臓病専門医や内分泌内科医へのコンサルテーションを行う。 |
| 摂食障害 |
|
構造的異常(例えば、腸回転異常/無回転,Hirschsprung病,遅発性横隔膜ヘルニアなど)の有無を評価するための消化器内科医へのコンサルテーション |
| 免疫不全 | 感染に対する積極的治療 | 稀に、抗生剤の予防投与、免疫グロブリン静注療法、胸腺移植が必要になる。 |
| 免疫の正常化が確認されるまで放射線照射血液製剤の使用が推奨される。 | ||
| 自己免疫疾患 | 免疫専門医の推奨に従った治療 | |
| 低カルシウム血症 | 標準的手法に従ったカルシウム補給 | 長期にわたるカルシウム補給を受ける罹患者については、腎結石のリスクが高まるため、内分泌内科医や腎臓内科医への紹介 |
| 成長ホルモン分泌不全 | 内分泌内科医による標準治療 | |
| 眼奇形 | 眼科医の推奨に従った標準治療 | 早期介入プログラムもしくは学区を通じて行う地域の視覚障害者サービス |
| 難聴 | 補聴器が有用なことあり;耳鼻科医の手で行う。 | 早期介入プログラムもしくは学区を通じて行う地域の聴覚障害者サービス |
| 神経系 | 低カルシウム血症に起因するものではないてんかん発作については抗てんかん標準治療 | |
| 発達遅延 |
|
|
| 精神疾患 | 精神科医や臨床心理士の推奨に従った支援や治療 | 早期診断・早期治療により長期予後が向上する |
| 頸椎異常 | 整形外科医の推奨に従い、活動を制限 | 代替活動を推奨することで協力性が向上する。 |
| 腎奇形 | 腎臓内科医の推奨に従った手術や治療 | 手術前には心臓病専門医や内分泌内科医への紹介が必要。 |
| エナメル質形成不全ないし齲蝕の多発 | シーラントも考慮しつつ、通常の治療を行う。 |
定期的追跡評価
表7:22q11.2欠失症候群罹患者に対して推奨される追跡評価の内容
| 系/懸念事項 | 評価 | 実施頻度 |
|---|---|---|
| 耳鼻咽喉系 | スピーチの鼻音化状態の評価 | 鼻咽腔閉鎖機能不全のスクリーニングを目的として、言語表出開始以降に行う。 |
| 免疫系 | 抗体陽転の確認を目的とした抗体検査 | 生後9ヵ月から12ヵ月の間に行う。 生ワクチン投与前には免疫の評価を繰り返し行うことを考慮する。 |
| 白血球百分率を含む全血算(CBC) | 年に1度 | |
| 内分泌系 | 血清カルシウム |
|
| 甲状腺刺激ホルモンならびに遊離T4 | 年に1度 | |
| 眼 | 眼科的評価 | 1歳から3歳の間、もしくは症候の状態に応じて適宜 |
| 聴覚 | 聴覚評価 | 乳児期、就学前、学齢期にそれぞれ行う。 |
| 発達 | 発達評価 | 年に1度 |
| 筋骨格系 | 脊柱側彎に関する臨床的追跡評価 | 年に1度 |
| 歯 | 歯科的診査 | 6ヵ月ごと |
避けるべき薬剤/環境
リンパ球に異常を有する乳児については、生ワクチン(経口ポリオ、MMRなど)の接種は避けるべきである。
炭酸飲料やアルコール飲料は、低カルシウム血症を悪化させる可能性がある。
カフェイン摂取により、不安を惹起ないし増悪させる可能性がある。
リスクを有する血縁者の評価
22q11.2DS罹患者の無症状の同胞ならびに両親については、可能な限り早期に心臓や免疫系の検査を受けたり、22q11.2DSに伴うその他の症候についての評価や追跡調査を受けたりすることができるよう、遺伝学的状態を明確にしておくことが望ましい。
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
妊娠に関する管理
妊婦については、その人のもともと有していた先天性心疾患、脊柱側彎、反応性気道疾患などの状態を念頭に、医学的モニタリングを必ず行う必要がある。これ加えて追跡すべき項目としては、カルシウム・甲状腺・血小板の値などがある。さらに、精神状態や行動状態に変化がみられるようなときは、メンタルヘルスを担う医療機関へのコンサルテーションを即座に行う必要がある。
22q11.2DSに関して高リスクの胎児については、先天性心疾患、羊水過多をきたす原因となりうる気道・口蓋・嚥下・消化器系の変化(先天性横隔膜ヘルニア・気管食道瘻・声門下狭窄・血管輪・喉頭横隔膜症・口蓋裂・口唇口蓋裂)、腎奇形、内反足や頭蓋縫合早期癒合症などの骨格変化、臍ヘルニアや鼠径ヘルニアといったもろもろの異常を調べるため、レベル2の胎児心エコー検査を行うべきである。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「ClinicalTrials.gov」、ならびにヨーロッパの「EUClinicalTrialsRegister」を参照されたい。
注:本疾患に関する臨床試験が掲載されているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
22q11.2欠失症候群(22q11.2DS)常染色体顕性の遺伝形式をとる隣接遺伝子欠失症候群である。
家族構成員のリスク
発端者の両親
- 3.0(2.54)Mbの欠失によって生じる22q11.2DSについては、罹患者の90%以上がdenovoの例で、ヘテロ接合体の親からの継承例は約10%である[Bassettら2011,McDonald-McGinnら2015]。
- 入れ子型欠失(すなわち、大きな定型欠失領域の内部で現れる反復性、非定型性のより小さな領域の分節的欠失[低コピー反復配列22B-D部])の罹患者については、親からの継承例の割合が高い(60%)。
- 再発危険率の判定の信頼性を担保するため、発端者の両親については、発端者で確認された欠失の有無を確認するためのゲノム検査が推奨される。
- 22q11.2の欠失が両親いずれの白血球DNAからも検出されなかった場合は、発端者に現れたdenovoの欠失、もしくは片親の生殖細胞系列モザイクのいずれかである。実際に、親の生殖細胞系列モザイクの例が報告されている[McDonald-McGinnら2015]。
- 臨床的に現れる症候にばらつきがある、あるいは浸透度が100%でないといった理由で、家族に22q11.2DSの症候がみられず、そのため一見したところでは家族歴がないように見受けられるような例もある。したがって、発端者の有する欠失が両親のいずれにもみられないことが検査で確認されない限り、家族歴が陰性であると確定的に言うことはできない。
- 注:仮に、発端者より先に片親のほうが病的バリアントを有していたということであれば、その片親が体細胞モザイクで、そのために症状が軽度ないし軽微となった可能性が考えられる。実際に、見かけ上、無症候の体細胞モザイクの成人の存在が確認されている[McDonald-McGinnら2015]。
発端者の同胞
発端者の同胞の有するリスクは、発端者の両親の臨床的状況や遺伝学的状態により変わってくる。
- 片親が罹患者、もしくは22q11.2の欠失をもつ場合は、同胞への再現危険率は50%である。22q11.2DSの臨床症状の現れ方には家系内で大きな幅がみられることから、この欠失を同胞が継承した場合に出現する表現型については予測不能である。
- 発端者の有する22q11.2の欠失が、両親いずれの白血球DNAからも検出されなかった場合、同胞への再現危険率は一般集団における出現率より若干高い程度となる。それは、片親の生殖細胞系列モザイクの可能性が考えられるからである[McDonald-McGinnら2015]。
- 両親が22q11.2欠失の検査を受けておらず、しかも臨床的には無症候であるといった場合であれば、発端者の同胞に再現する確率は低いものと思われる。しかし、両親が臨床的にみて罹患者ではないと思われる場合でも、片親がヘテロ接合で浸透率が100%でなかった可能性、あるいは片親の生殖細胞系列モザイクの可能性があるため、22q11.2DSの再現危険率は通常より高くなるものと思われる。
- 可能性は低いながら、片親が22q11.2領域を含む均衡型の染色体再配列を有している場合は、同胞への再現危険率は高まることになる。具体的な再現危険率の数値は、染色体再配列の内容によって異なる。
発端者の子
22q11.2DS罹患者の子については、いずれも、22q11.2欠失を継承する可能性が50%となる。
他の家族構成員
他の血族の有するリスクは、発端者の両親の状態によって変わってくる。仮に片親も欠失を有していたということになれば、その血族にあたる人はすべてリスクを有することになる。
関連する遺伝カウンセリング上の諸事項
家族計画
- 遺伝学的リスクの確定、出生前検査/着床前検査を受けるかどうかの話し合いに最も適しているのは、妊娠前の時期である。
- 罹患者である若い成人、リスクを有する若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
DNAバンキングとは、将来、使用できるようになった場合に備えてDNA(典型的には白血球から抽出したDNA)を保存しておくことをいう。検査の手法や、遺伝子・遺伝子のバリアント・疾患等に対するわれわれの理解が、将来はより進歩していくことが予想される。そのため、罹患者のDNAについては、保存しておくことを考慮する必要がある。
出生前検査ならびに着床前遺伝学的検査
分子遺伝学的検査
高リスクの妊娠
- 分子遺伝学的検査
血族の1人に22q11.2の欠失の存在が確認された場合には、FISHやMLPAを用いて、高リスク妊娠に関する出生前検査や、着床前遺伝学的検査を行うことが可能となる。
- 超音波検査
高リスクの妊娠については、妊娠18週から22週の間に、口蓋関連異常を調べるための高解像度超音波検査、ならびに心奇形を調べるための心エコー検査を行うことが可能である。
注:妊娠週数とは、最後の月経の初日から数えた月経週数、もしくは超音波での計測値をもとに算出したものをいう。
低リスクの妊娠
家族歴からは22q11.2DSに関して高リスクと思われない妊娠においても、超音波検査で先天性心疾患や口蓋裂・口唇口蓋裂がみられる場合は、22q11.2DSが示唆される。特に、大動脈弓離断・総動脈幹・Fallot四徴・心室中隔欠損といった円錐動脈幹奇形を呈する例についてはそれが言える。22q11.2DSにおいてみられ、出生前の段階で把握可能なその他の構造的異常としては、先天性横隔膜ヘルニア、臍ヘルニア/鼠径ヘルニア、気管食道瘻/食道閉鎖/喉頭閉鎖、多指趾、頭蓋縫合早期癒合症、多小脳回、腎奇形などがある。それに加え、羊水過多もしばしばみられる。胎児の細胞から抽出した染色体をアレイ検査、MLPA法、FISH法で分析することが可能である。22q11.2DSの診断確定が仮に妊娠後期になったとしても、周産期の管理のことを考えると、十分意味があると言える。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- International22q11.2DeletionSyndromeFoundation,Inc.
P.O.Box2269
CinnaminsonNJ08077
Phone:877-739-1849(toll-free)
Email:info@22q.org
www.22q.org
- MaxAppeal
15MeridianAvenue
StourbridgeWestMidlandsDY81049
UnitedKingdom
Phone:08003891049tollfree
Email:info@maxappeal.org.uk
www.maxappeal.org.uk
- My46TraitProfile
22q11.2 Deletion Syndrome
- NationalLibraryofMedicineGeneticsHomeReference
22q11.2 deletion syndrome
- NCBIGenesandDisease
DiGeorge syndrome
- Chromosome22Central
c/oMurneyRinholm
7108PartinwoodDrive
Fuquay-VarinaNC27526
Phone:919-567-8167
Email:usinfo@c22c.org
www.c22c.org
- ChromosomeDisorderOutreach(CDO)
POBox724
BocaRatonFL33429-0724
Phone:561-395-4252(FamilyHelpline)
Email:info@chromodisorder.org
www.chromodisorder.org
- MedicalHomePortal
MedicalHomePortalの「親と家族」のセクションでは、家族が、慢性的で複雑な疾患を抱える子どものケアをより良く行う方法を学ぶとともに、子どものケアにおいてより優れたパートナーになる方法を学ぶための情報や資源を提供している。
DepartmentofPediatricsUniversityofUtah
P.O.Box581289
SaltLakeCityUT84158
Phone:801-213-3920
Email:info@medicalhomeportal.org
ForParents&Families
- EuropeanSocietyforImmunodeficiencies(ESID)Registry
Dr.GerhardKindle
UniversityMedicalCenterFreiburgCentreofChronicImmunodeficiency
Engesserstr.4
79106Freiburg
Germany
Phone:49-761-270-34450
Email:esid-registry@uniklinik-freiburg.de
ESID Registry
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:22q11.2欠失症候群:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specific データベース |
HGMD | ClinVar |
|---|---|---|---|---|---|
| 対象外 | 22q11.2 | 対象外 | |||
| TBX1 | 22q11.2 | Tボックス転写因子TBX1 | TBX1database | TBX1 | TBX1 |
データは、以下の標準資料から作成したものである。遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:22q11.2欠失症候群関連のOMIMエントリ(OMIMへ)
| 145410 | none found |
| 188400 | DIGEORGE SYNDROME; DGS |
| 192430 | VELOCARDIOFACIAL SYNDROME; VCFS |
| 600594 | DIGEORGE SYNDROME CRITICAL REGION GENE 2; DGCR2 |
| 601279 | DIGEORGE SYNDROME CRITICAL REGION GENE 6; DGCR6 |
| 601755 | ESS2 SPLICING FACTOR, XENOPUS, HOMOLOG OF; ESS2 |
| 602054 | T-BOX TRANSCRIPTION FACTOR 1; TBX1 |
| 609030 | DGCR8 MICROPROCESSOR COMPLEX SUBUNIT; DGCR8 |
分子レベルの病原
22q11.2の3.0Mbの欠失(CMA上は、UCSCゲノムブラウザ、GRCh37塩基配列座標系の18,912,231-21,465,672の2.54Mbの欠失という表記になっている)により、いくつかの関連遺伝子が欠失することになる(この後に続く「この領域における注目遺伝子」の項を参照のこと)。
22q11.2欠失症候群の罹患者の85%以上は、同じ3.0Mb近く(CMAでは2.54Mb)の領域の欠失を示す。残りの罹患者は、欠失終点のバリアント、あるいは、大きな定型欠失領域(TDR)内で小さな欠失セグメントが入れ子状に散在する反復性・非定型性の欠失のいずれかである(図1参照)[Levyら1995,Kurahashiら1996,O‘Donnellら1997,McQuadeら1999]。
- 3.0(2.54)Mbの欠失:ISCN-37446
- 2Mbの非定型欠失:ISCN番号なし
- 1.5Mbの欠失:ISCN-37516
- 遠位欠失:ISCN-46292
典型的な軟口蓋心臓顔面症候群/DiGeorge症候群の表現型を示す1患者で、TDR内の20kbの微小欠失がみられた例の報告がみられる[Yamagishiら1999]。この微小欠失では、UFD1LとCDC45Lが影響を受ける。その他、定型的な欠失と部位が重ならず、それより遠位の点からテロメアに向かう欠失を示す例も数例存在する。22q11.2の欠失終点の近傍に重複配列のブロックがあることで、定型的・非定型的欠失に至ることが強く示唆される。
不均衡型転座の結果として、22pter→q11領域の欠失を示す罹患者の例が少数みられる(詳しくは表Aの「Locus-Specificデータベース」の欄を参照のこと)。
この領域における注目遺伝子
- TBX1
TBX1の欠失は、先天性心疾患に関係してくる(「鑑別診断」の「単一遺伝子疾患」の項を参照のこと)。それに加え、TBX1は、心臓・咽頭・脳の微小血管や、認知・行動の障害にも関連してくる[McDonald-McGinnら2015]。
- DGCR8
DGCR8は、エピジェネティックな役割を果たしているものと思われ、22q11.2DSの表現型における精神神経学的側面その他に寄与する遺伝子の発現を修飾しているようである。
- CRKL
CRKLは心奇形に関係すると考えられており、ナチュラルキラー細胞の機能を調節しているようである。
- SNAP29
SNAP29の病的バリアントは、潜性遺伝疾患であるCEDNIK症候群(OMIM609528)に関連するとされている。
- PRODH
PRODHの病的バリアントは、潜性遺伝疾患である高プロリン血症Ⅰ型(OMIM239500)に関連するとされている。
更新履歴:
-
Gene Review著者: Donna M McDonald-McGinn, MS, CGC, Beverly S Emanuel, PhD, FACMG,
and Elaine H Zackai, MD, FACMG
日本語訳者: 末國久美子,山本佳世乃,川目裕(お茶の水女子大学大学院人間文化創成科学研究科ライフサイエンス専攻遺伝カウンセリングコース)
Gene Review 最終更新日: 2005.12.16. 日本語訳最終更新日: 2012.6.25. - Gene Reviews著者: Donna M McDonald-McGinn, MS, LCGC, Heather S Hain, PhD, LCGC, Beverly S Emanuel, PhD, and Elaine H Zackai, MD, FACMG
日本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)GeneReviews最終更新日:2020.2.27. 日本語訳最終更新日:2024.4.7.[in present]
![]()