先天性副腎過形成 21水酸化酵素欠損症
(21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia)
[同義語: 21-OHD; CAH, 21-OHD; Virilizing Adrenal Hyperplasia]
Gene Reviews著者: Saroj Nimkarn, MD, Prasanna K Gangishetti, MBBS, Mabel Yau, MD, and Maria I New, MD.
日本語訳者:和泉 賢一 (札幌医科大学医学部遺伝医学,NGSDプロジェクト)
Gene Reviews 最終更新日: 2016.2.4 日本語訳最終更新日: 2017.12.29
原文 21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia
要約
疾患の特徴
I21水酸化酵素欠損症(21-OHD)は先天性副腎過形成(CAH)のなかで最も頻度の高い疾患であり,副腎皮質におけるコレステロールからコルチゾールへの合成障害を認める常染色体劣性遺伝疾患である.21-OHD CAHでは,副腎アンドロゲンの過剰な生合成によりすべての患者に男性化を認め,また一部の患者に塩類喪失を認める.重症の酵素欠損を認め出生前に発症する古典型と,軽度の酵素欠損を認め出生後に発症する非古典型とに区別される.古典型はさらに単純男性化型(~25%)とアルドステロン産生が不十分な塩類喪失型(75%以上)に分けられる.塩類喪失型の21-OHDの新生児は生命を脅かす塩類喪失クリーゼのリスクがある.非古典型の患者は中等度の酵素欠損のみであり,出生後に高アンドロゲン血症を示す;非古典型の女性は出生時には男性化は認めない.
診断・検査
古典的21OHD CAHの診断は,新生児期では,血清17-OHPの上昇と副腎アンドロゲンの上昇という臨床的特徴で確立されている.
非古典的21-OHDの診断は,ベースラインの血清17‐OHPとACTH負荷時の血清17-OHPの比,もしくは早朝時の17-OHPの上昇によって行われる.両アレルの病的CYP21A2変異を認めれば、臨床診断が確定され,家族の診断も可能になる.
臨床的マネジメント
症状による治療:
古典的21-OHD CAH:グルココルチコイド補充療法であり、ストレス下では増量する. 塩類喪失型:ミネラルコルチコイド 9α‐fludrohydrocortisoneとしばしば塩化ナトリウムを使用する.生下時に男性化を認めている女児は女性生殖器形成術とともに,もしくは膣拡張術を必要とすることがある.症状のある非古典的21-OHD CAHは治療を必要とすることがある.
初期症状の予防:
新生児スクリーニングプログラムは,潜在的に生命にかかわる塩類喪失クリーゼの前にグルココルチコイドとミネラルコルチコイド補充療法を開始するため,古典的21-OHD CAHの児を同定する目的で行われる.
サーベイランス:モニター:
- 元気に子供が成長している間は3-4か月ごと,その後はもう少し頻度を落とす,
- 男性の精巣由来副腎残存腫瘍は思春期が始まったあとは3年から5年ごと;
- 体重,骨塩濃度,生殖能,心血管,メタボリック症候群のリスクが大人にはある.
リスクのある血縁者の評価:
早期診断・早期治療を容易にするために,リスクのある兄弟姉妹の17‐hydroxyprogesterone (17-OHP)を測定するのは適切である.遺伝カウンセリング
21-OHDは常染色体劣性遺伝疾患である.ほとんどの親は一方が正常,もう一方が変異アレルというヘテロ接合体である.約1%の変異は新生突然変異で起こる.したがって発端者の1%は片方の親のみがヘテロ接合体である.またそれまで罹患しているとは思われていなかった親が両アレル変異をもつ非古典的21-OHDであると判明するケースもある.もし発端者の両親がいずれもヘテロ接合体で変異がある場合,変異アレルを両方受け継いで罹患する確率は25%,無症候性キャリア(片方が変異アレルのヘテロ接合体)が50%,罹患せずキャリアでもない確率が25%である.リスクのある血縁者の保因者診断と児の出生前診断は,家系内での病的変異が分かっていれば可能である.
GeneReview Scope
| 21‐水酸化酵素欠損先天性副腎過形成: 表現型を含む |
|---|
|
同義語や旧名はGeneReview本文のNomenclature参照.
診断
疑うべき所見
21-水酸化酵素欠損症による先天性副腎過形成(21-OHD CAH)は以下の場合本疾患を疑う.
- 出生時に男性化を認める女児,出生後に男性化を呈する女児,
思春期早発や副腎皮質性思春期徴候を認める女児.男性化は成熟,成長(高身長に至る),性ホルモン感受性部位(外性器,皮膚,髪)(2次性徴に至る)に影響を及ぼす. - 小児期に男性化を認める男児(偽性思春期早発症)
- 生後4週以内に塩類喪失クリーゼを起こした新生児.治療していない,または,塩類がコントロールできない人は,血清Na濃度,Cl濃度,CO2濃度の低下,血清Kの増加,尿中Na濃度の異常な増大が起きる.
- 新生児スクリーニング陽性として発見された17-OHP濃度増加の幼児
注:21-OHD CAHの女児は46,XXの核型をもつ.21-OHD CAHの男児は46,XYの核型をもつ.
新生児スクリーニング
21-OHDの新生児スクリーニングには2つの目的がある.
- 生命を脅かす塩類喪失クリーゼのリスクのある古典型21-OHDの乳児を見つけること
- 外性器形態があいまいな女児の診断を円滑に進めるため
注:新生児スクリーニングは非古典型21-OHDの患者を検出することはほとんどない [Votava et al 2005].
21-OHDの新生児スクリーニングが義務づけられている州はNational Newborn Screening Status Report(pdf)で確認できる.17-OHPの濃度測定はほかの新生児スクリーニングで用いられているのと同じように踵より採血し,ろ紙に採取した血液スポットで行われる.
- 多くのスクリーニングプログラムは単回の測定を行っており17-OHP濃度に疑問のもたれる結果に対する再検査は行っていない.Speiser et al [2010] 原文参照.
- スクリーニングプログラムによっては,スクリーニングの有効性を改善する目的で,スクリーニングで境界領域だった1次検査の結果と2次検査の結果を再検討し,施設によってスクリーニングを再度行うところもある[Sarafoglou et al 2012, Chan et al 2013].免疫学的検査の偽陽性率は高いため,2回目の検査として液体クロマトグラフィータンデム質量分析法を推奨している[Speiser et al 2010].1次検査陽性のサンプルに種々のホルモン(17-OHP,Δ4-androstenedione,コルチゾール)の測定を2次検査として行っているプログラムもある[Janzen et al 2010].州によっては,精度を上げるため,乾燥血液スポットの免疫アッセイの前に有機溶媒抽出を義務付けているところもある.
注:(1)生後24時間以内に採取された検体では全ての新生児で値は上昇しており,偽陽性結果を示すことがある.(2)偽陽性結果は低出生体重児や未熟児でも認められる.それゆえ,もし,検査が陽性であったときは,生下時体重または妊娠週数で補正された標準データを診断に用いる. (3)関係ない問題でデキサメサゾンを投与された新生児においては偽陰性が認められる.
確立された診断法
21-OHD CAH. 新生児の確定診断は,次のような検査結果で行われる.
- 血清17-OHP は著明に上昇する.
- 副腎アンドロゲンは上昇する;Δ4-androstenedione, 21 deoxycortisolそしてプロゲステロンが21-OHD CAHの男性と女性で上昇する;テストステロンと副腎アンドロゲン前駆体(Δ4-androstenedione, DHEA)は罹患した女性と思春期前の男性で上昇する.
- 血漿レニン活性(PRA)は、21-OHD CAHの塩類喪失型の患者で著明に上昇する
注:塩類喪失型の21-OHD CAH患者においては,血清中のアルドステロン濃度はPRAの上昇の程度に比して不適切に低値となる.PRAに対する血清アルドステロンの比の減少はアルドステロン合成障害を示し,新生児期後にてCAHの単純男性化型と塩類喪失型の鑑別が可能である [Nimkarn et al 2007].
非古典型 21-OHD CAH.発端者の診断法は2つの臨床検査法の一つでの結果を基に確定される(Figure 1とTable 1を参照).
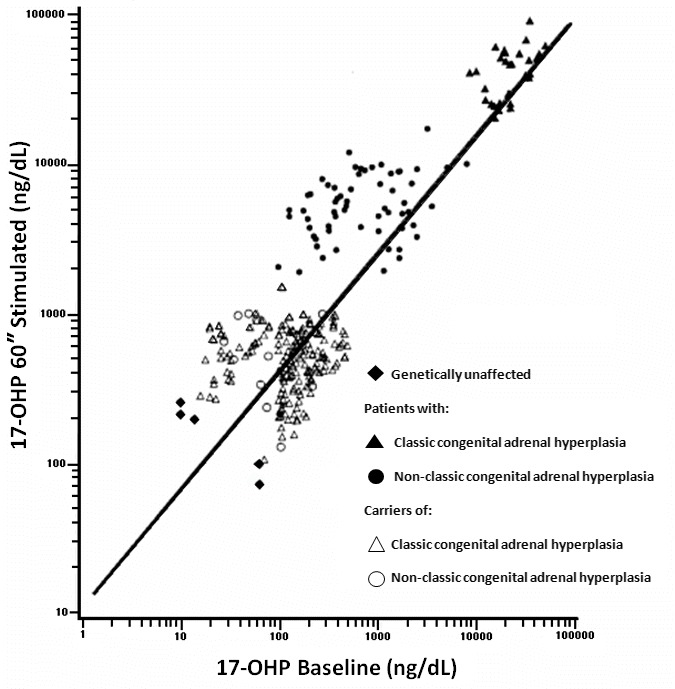
図1. 21-OHD CAHの診断のための17-OHP 計算図表(60分コートロシンTM 負荷試験).
- 60分ACTH負荷試験. 250μgの合成ACTH(コートロシンTM)経静脈的にボーラス注入した後,60分後測定した17-OHPの血清濃度をFigure 1の計算図表に記入する.
- 17-hydroxyprogesterone (17-OHP).単回早朝(午前8時前)の17-OHP濃度測定(罹患者の基礎値は常に上昇しているわけではない;Table 1を参照)
注:17-OHPの思春期および性別による基準値は,行っている検査法を反映して検査会社によって異なっている.成人女性では、基準値は、生理周期の時期に影響される.
表 1. 幼年期以降の17 OHPレベルによる21-OHD CAHの診断
| 古典型 | 非古典型 | 罹患していない人 | |
|---|---|---|---|
| 17-OHP 基礎値 | >10,000 ng/dL or 300 nmol/L | 200-10,000 ng/dL or 6-300 nmol/L1 | <200 ng/dL or 6 nmol/L1 |
| ACTH 負荷後17-OHP値 | >10,000 ng/dL or 300 nmol/L | 1,000-10,000 ng/dL or 31-300 nmol/L | <1,000 ng/dL or 50 nmol/L |
Spiserらの表を改変[2010]
1.非古典的21-OHD CAHの場合,ランダムに測定した17‐OHPは基準値内のことがある.
分子遺伝学的検査
CYP21A2(Table 2を参照)の両アレルの病的変異の同定で、診断は確定し、家族の検査が可能になる.分子学的検査アプローチは,単一遺伝子検査,多遺伝子パネル使用,およびもっと包括的なゲノム検査を含んでいる.
- 単一遺伝子検査.CYP21A2遺伝子のシークエンス解析は最初に行われ,病的変異が一つもしくは病的変異が認められなかった場合,引き続き欠失・増幅解析が行われる.
注:広範囲な遺伝子変換(分子遺伝学参照)によって,機能のあるCYP21A2シークエンスの広い領域が,一つ以上の欠失変異の結果で機能を持たなくなったCYP21A1P偽遺伝子の領域に置き換わっていることがある[Mao et al 2002].このように,標的解析がたくさんの病的変異を発見したとき,病的変異はトランス型(すなわち、どちらの親からもうけついだもので,別々の染色体上にある)か,シス体(どちらかの親から遺伝子変換によって起こった可能性が最も高いであろう2つ以上の変異のある1つの変異アリルを受け継いだもので,同じ染色体上にある)かのいずれかである.誤診を避けるため,発端者と同様に両親の解析も,変異がシス体かトランス体かを確定し、病的変異を確定するため行うことが勧められる.
- 多遺伝子パネルはCYP21A2と他の関連ある遺伝子(鑑別診断の項を参照)を含むものであり、これも検討をされるかもしれない.注:研究室や時間により、多遺伝子パネルの感度および含まれる遺伝子の感度は様々である.多遺伝子パネルについての更なる情報はGene Reviewのページよりサイト移動ができる.
- 包括的ゲノム検査(利用できる)はエキソームシークエンス,ゲノムシークエンス、ミトコンドリアシークエンスを含んでおり,もし単一遺伝子検査(および,または多遺伝子パネル検査)で21 -OHD CAHの症状をもつ患者の診断が確定できないときは、検討されることがある.
表 2. 21-OHD CAHの分子遺伝学的検査
| Gene1 | 検査法 | この方法による病的変異2をもつ発端者の割合 |
|---|---|---|
| CYP21A2 | シークエンス解析法3 | ~70-80%4 |
| 標的遺伝子欠失/増幅解析法5 | ~20-30%6 |
- Table A参照
- この遺伝子のアリル変異の情報は分子遺伝学の項目参照
- シークエンス解析はbenign, likely benign, of uncertain significance, likely pathogenic, pathogenicの変異を検出する.Pathogenic(病的)変異は,遺伝しない欠失/挿入,ミスセンス,ナンセンス,スプライスサイト変異を含む;通常はエクソンまたは全ゲノム欠失/増幅は検出できない.シークエンス解析の解釈については,本文より参照ページへ.
- 21-OHD CAHのヘテロ変異の多くは、複合ヘテロである[Krine et al 2000, New et al 2013].
- 標的遺伝子欠失/増幅解析は遺伝子内欠失または増幅を検出する.次のような方法がある.
定量的PCR,広範囲PCR,multiplex ligation-dependent probe amplification (MLPA)法,1つのエクソンの欠失または増幅を検出するデザインの標的遺伝子マイクロアレイ. - 変異アレルの約20%がCYP21A1P偽遺伝子,アジュバンドC4B遺伝子の3末端かCYP21A2の5末端の領域を含む30kb遺伝子領域で検出される.
臨床像
臨床症状
先天性副腎過形成(21-OHD CAH)は古典型としても非古典型としても現れうる(表 3).
古典型21-OHD では出生前の重要な性分化の重要な時期にテストステロンやΔ4 -アンドロステンジオンなどのアンドロゲンに曝露される結果,遺伝学的には女性である患者の外性器の男性化をきたし,時には出生時に不明瞭な外性器形態を伴う.古典型はさらに単純男性化型(~25%)とアルドステロン合成が不十分な塩類喪失型(~75%)に細分される.21-OHD によって塩類喪失型CAHを生じている新生児では生命にかかわる塩類喪失クリーゼをおこす危険がある.
非古典型21-OHD の患者の症状は中等度酵素欠損のみで,出生後にアンドロゲン過剰症状を示す.非古典型の女性患者では出生時の男性化はみられない.
表 3. 古典型,非古典型21-OHD患者の臨床像
| 徴候 | 21-OHD | |
|---|---|---|
| 古典型 | 非古典型 | |
| 出生前男性化 | 女性に見られる | なし |
| 出生後男性化 | 男女とも見られる | さまざま |
| 塩類喪失 | 全患者の~ 75% | なし |
| コルチゾール欠損 | ~100% | まれ |
古典型単純男性化型 21-OHD
副腎アンドロゲン合成過剰. 子宮内での合成過剰は 46,XXの正常核型をもつ女児の出生時の男性化を招く.罹患女性ではアンドロゲン過剰によりさまざまな程度の陰核肥大,陰唇融合,尿道膣瘻の形成を招く.抗ミュラー管ホルモン(AMH)が分泌されないため,罹患女性のミュラー管から正常に子宮と卵管は形成される.罹患女児の出生時の男性化の程度からのみでは単純型21-OHDと塩類喪失型21-OHDを鑑別することはできない.
出生後,グルココルチコイド補充を受けない単純男性化型 21-OHD患者では男女とも陰毛や腋毛の早期出現,座瘡,急速な身長の伸び,骨年齢の促進といったアンドロゲン過剰による徴候が現れる.無治療の男性では陰茎の肥大と小さな精巣を認める.無治療の女性では陰核肥大,多毛,男性型頭部脱毛,月経異常,妊孕性の低下を認める.
無治療の21-OHD患児では当初の身長の伸びは急速であるが,骨端の早期閉鎖のために成人期の最終身長は低くなる.コルチゾール補充による治療を早期に開始し副腎アンドロゲン分泌をコントロールできたとしても,通常患者は期待する最終身長には到達しない.骨年齢は実年齢より促進している.
思春期の発来. グルココルチコイドの治療を行われ副腎アンドロゲンの産生を抑制された児は男女ともにたいていは適切な年齢で思春期が発来する.しかし,疾患がよくコントロールされている患者の中でも例外はありうる[Trinh et al 2007].
早期に治療を行われずにいた子供たちは,グルココルチコイドの補充療法を開始することが思春期早発のトリガーとなることに留意が必要である.これは副腎アンドロゲンの大量分泌に由来するエストロゲンが視床下部下垂体系を抑制しておりグルココルチコイド治療を行った場合に,この抑制が解除されることで中枢性の思春期早発が起こると考えられる.
妊孕性. 適切に治療された女性の大部分は初潮後の月経は正常で妊娠も可能である[Lo et al 1999].しかし,妊娠率は低いと報告されている.その理由として,満足の得られる性交ができない不十分な膣口例,破瓜の痛み[Gastaud et al 2007]やアンドロゲンの上昇により卵巣機能が低下している例,性別やセクシャルパートナーの選択に関する性心理の問題も含まれているためである.慢性排卵障害,プロゲスチンレベルの上昇,異常子宮内膜移植が低妊孕性の理由として挙げられている[Wichel 2012].
男性の生殖能低下の主な原因は精巣副腎残存腫瘍 (testicular adrenal rest tumor; TART) が存在することである.それは異常な副腎組織に由来すると考えられている.さらに,低ゴナドトロピン性性腺機能低下症は副腎アンドロゲンや代謝産物の過剰によって下垂体のLH分泌が抑制されることによる[Ogilvieet al 2006a].
副腎髄質. 古典的 21-OHD CAH患者ではコルチゾールの欠乏が成長や副腎髄質機能にも影響を与え,非罹患者に比べエピネフリンやメタネフリンのレベルは低くなる[Merke et al 2000].
古典的塩類喪失型 21-OHD CAH.21-OH機能の喪失が重度である場合,腎遠位尿細管からのナトリウム再吸収に必要な副腎アルドステロン分泌が不十分となり,患者はコルチゾール欠乏やアンドロゲン過剰とともに塩類喪失も伴う.腎性塩類喪失を伴う新生児は哺乳不良,体重減少,成長不全,嘔吐,脱水,低血圧,低ナトリウム血症,高カリウム血症を示し,副腎クリーゼ(高窒素血症,循環不全,ショック,死亡)に進展する.副腎クリーゼは早ければ出生後1から4週におきてくる.
外性器の形態からは認識されないため,新生児スクリーニングで発見されなかった男児は塩類喪失型副腎クリーゼの高いリスクを有する.こうした患者はしばしば診断されないまま退院し,家で塩類喪失クリーゼを経験することになる. 逆に性器形態があいまいな塩類喪失型の女児は早期に診断され,治療を受けることができる.
明らかな塩類喪失クリーゼは子供を塩類喪失型と診断させるが、ある程度のアルドステロン欠乏は、レニン負荷アルドステロン産生能によって判断され,21 OHD CAHとして判明する[Nimkarn et al 2007].
非古典型 21-OHD CAH
非古典型 21-OHD CAHは出生後どの時期にも座瘡,陰毛の早期出現,急速な成長,骨年齢の促進,さらに古典型21-OHD CAHと同様早期骨端閉鎖による最終的な低身長,といったアンドロゲン過剰による症状を伴って発症する[New 2006].軽度のコルチゾール合成能の低下は非古典的21-OHD CAHの患者では臨床的に明らかではない.
非古典型21-OHD CAHに罹患した女性. 罹患児の出生時の外性器の男性化を推察するのは困難である[Kashimada et al 2008].罹患児は出生時は通常の性器で生まれる.出生後に多毛,側頭部の脱毛,初経の遅れ,不規則な月経,不妊といった症状を呈してくる.21-OHD CAHの成人女性の約60%は多毛を示すのみで,約10%が多毛と月経異常,そして約10%は月経異常のみを示す.多くの非古典型の女性は多嚢胞性卵巣を生じる.高アンドロゲン血症を示す女性の2.2‐10%に非古典型21-OHD CAHが同定される[NEW 2006, Escobar-Morreale et al 2008. Fanta et al 2008].無治療女性の妊孕性は50%と報告されている[Pang 1997].
非古典型21-OHD CAHに罹患した男性. 非古典型21-OHD男性は早期にあごひげが生えたり,比較的小さい精巣に大きな陰茎を伴ったりする. 一般的に性機能不全は認めない.精子の数も正常である[New 2006].両側の副腎皮質偶発腫は非古典型CAHの成人男性に報告された唯一の所見である[Nigawara et al 2008].
性別と行動. 古典型21-OHDの女児は出生前のアンドロゲン暴露によって外性器形態と幼少期の態度に影響を及ぼす.幼少期の遊技行動は女性としてのジェンダーの減弱に関与し,成人してからの異性愛への興味を減弱させる.罹患女性はジェンダーに対して身体的違和感をより覚えるようになるが異性愛への興味は減弱する傾向を示す.女性という性別を割り当てられることに対する満足感が少ない.逆に21-OHDの男性は幼少期の行動やジェンダーの認識,性的立場に大きな変化は見られない.出生前のアンドロゲン暴露は成人女性としての女らしさを減弱させるが,男らしさを増加させるわけではない[Long et al 2004].
両性と同性の性的嗜好の割合は,どの分類の21-OAD CAHの女性でも増加しているが,出生前のアンドロゲン暴露の度合いに相関することがわかった.両性/同性嗜好の割合は,非性的行動での全体的な男性化の程度に関連し,出生前アンドロゲン暴露と子供時代の行動の男性化により,独立に予測される[Meyer-Bahlburg et al 2008]. 逆に,21-OHD
CAHの男性には子供時代の行動,性同一,性的嗜好に一般的な変化は認められない[Hines et al 2004].
病因. 21-水酸化チトクローム450の機能が不十分であると,コルチゾール産生経路がブロックされ,17-OHPが蓄積される.17-OHPが過剰になるとアンドロゲンの経路へ流れ,17,20-lyaseにより17-OHPはΔ4-アンドロステンジオンに変化し,それがアンドロゲンとなる.ミネラルコルチコイドの経路は21-ハイドロオキラーゼの活性がわずかでも必要であるため,ミネラルコルチコイド欠損(塩類喪失)はこの疾患の最も重症型を呈する.
ステロイド合成の欠如は下垂体からのACTH分泌に対してネガティブフィードバック機能を損なうため慢性的なACTHによる副腎皮質刺激を引き起こし副腎過形成となる.
遺伝子型‐表現型の相関
21‐OHD CAH罹患者の大きなコーホート研究を含むNew et al [2013]による研究にて、考えられてきたより表現型の発現率が少ないことがわかった.直接的な遺伝型‐表現型の相関は遺伝型の約50%であった.関係が最も信頼できない型は単純男性型で,同じ遺伝子型を示しても、表現型には多くの幅があった.しかし,塩類喪失型と非古典型では,いくつかの遺伝子型で強い相関が独占的に認められた.例えばVal281Leu病的変異はもっぱら非古典型と相関している.この型は,表現型の効果は2つのアレルの重篤ではない病的変異を反映している.
アレルは残存酵素活性により重度と軽度に分けられる(Table 4).
- 塩類喪失型21-OHD CAHは通常最も重篤な病的変異である.(例えば、欠損のホモ接合)
- 非古典的21-OHD CAHは通常一方の軽度のアリルか両方の軽度のアリルである.
出生前診断のとき、生下時に治療を行う必要性を決定するため、古典的または非古典的遺伝子型を区別することは難しい.
- 発端者が男性化した女性の家族では、次に生まれる罹患した女性の胎児の男性化のリスクを予見するのは可能である.
- 発端者が男性の家族では、次に生まれる罹患した女性の胎児の遺伝子型の男性化のリスクを予見するのは難しい(信頼できない).
古典的 21-OHD CAH. 21-OHD CAHの古典型の遺伝子型はin vitroの実験で確かめられた完全に酵素活性が失われる重篤な病的変異が両アリル上にあることが予想される.
注:最も頻度の高い古典的21-OHD CAHの病的変異であるc.293-13A>Gまたはc.293-13C>Gの単一ヌクレオチド変異は,イントロンの不完全なスプライシングと翻訳リーディングフレームのシフト(フレームシフト)を引き起こす.この変異によるホモアリルをもつ患者のほとんど(90%)が塩類喪失型21-OHDであるが,塩類喪失の程度はさまざまである.遺伝子型と表現型が一致しないのは接合部位の変異により正常なスプライシングが行われず,互い違いにスプライシングされる部分が増加することによると説明できる.いくつかの蛋白は産生されるが,活性にばらつきが生じる[Higashi et al 1988].
病的変異であるp.Ile173Asnと次の重篤な変異をもつ複合ヘテロの罹患者の中では、76%が単純男性化21OHD CAHの表現型だが,23%は塩類喪失型の表現型を示す[New et al 2013].転写制御や下流の蛋白翻訳に関わる小変異は、おそらく21‐OH酵素活性を減少させるのであろう.
非古典的 21-OHD CAH. 非古典型 21-OHDCAH患者は両アレルに軽症型の変異を有しているか,1アレルに重症型変異,1アレルに軽症型アレルを有していると予測される.約2/3の非古典型21-OHD患者は複合ヘテロである.エクソン1(p.Pro31Leu)とエクソン7(p.Val282Leu)のミスセンス変異は酵素活性を減じ,非古典的21-OHDに関与する.しかし、一つの軽症型変異に関係した表現の多様性は認められる.
- p.Val282Leuかp.Pro31Leu病的変異や重篤な1変異をもつ患者の少数(<3%)で、非古典的表現型と考えられても、古典的表現型が認められることがある.
- p.Ile173Asn病的変異や重篤な1変異をもつ患者の非常に少数だが、(古典的が発症すると考えられる場合でも)非古典的表現型となることがある[Stikkelbroeck et al 2003].
表 4. 残存酵素活性による,よく認める CYP21A2 変異の分類
| 酵素活性 | 変異カテゴリー | 変異 |
|---|---|---|
| 0% | 重度(古典型) | 遺伝子全欠損(null変異),大きな遺伝子転換,p.Gly111ValfsTer21, p.[Ile237Asn;Val238Glu;Met240Lys], p.Leu308PhefsTer6, p.Gln319Ter, p.Arg357Trp |
| <1%1 | c.293-13 A>G c.293C>G |
|
| 2-11% | p.I173Asn | |
| ~20-50% | 軽度(非古典型) | p.Pro31Leu,p.Val282Leu,p.Pro454Ser |
Krone et al[2000] 1, 最小残存酵素活性
隣接遺伝子欠損. CYP21A2とTNXを含む隣接遺伝子欠損は、関節可動亢進型のエーラスダンロス症候群と21-OHD CAHの両者を引き起こす[Burch et al 1997, Schalkwijk et al 2001].
専門用語について
21-OHD CAHには以前は以下のものが含まれていた:副腎性器症候群(AG症候群),先天性副腎皮質過形成
非古典型21-OHD CAHは以前は軽度のあるいは遅発性の病型として言及されていた.
塩類喪失型21-OHD CAHはsalt-wasting CAHまたはsalt-losing CAHとよばれる.
疫学
古典型21-OHD CAH. 約650万人の異なる人種における新生児スクリーニングのデータ解析によれば古典型21-OHDの頻度は出生15,000人あたり1人である[van der Kamp & Wit 2004].
人種ごとの頻度
- アラスカのYupikエスキモーでは1/300
- サウジアラビアでは1/5,000
- ヨーロッパと北米が1/10,000-16,000
- 日本人では1/21,000
- ニュージーランドでは1/23,000
非古典型21-OHD CAH. 多彩な人種で構成されるニューヨーク市における非古典型の頻度は1/100と推定されている.非古典型21-OHDの頻度が最も高い人種はアシュケナジーユダヤ人で1/27である.その他頻度の高い人種集団はヒスパニック(1/40),スラブ人(1/50),イタリア人(1/300)である[Speiser et al 1985].
遺伝学的に関連した(アレル)疾患
このGeneReviewで述べられている以上の病的CYP21A2変異に関与した表現型はない.
鑑別診断
副腎皮質の束状層におけるコルチゾール生合成には 5つの主要な酵素による合成ステップをふむ.先天性副腎過形成(CAH)はこのいずれの酵素の欠損によっても生じる.コルチゾールの合成障害はACTHの慢性的な上昇を招き,これが副腎の過剰に刺激して過形成をきたす.CAHの5つの型をTable5に示す.副腎皮質の生合成のそれぞれのステップにおける酵素機能障害が前駆体を蓄積させ欠損物質が生じる.21-OHDはCAHの90%以上を占め,最も多い.
表 5. CAHをきたす酵素欠損
| CAH中の割合 | 欠損酵素 | 基質 | 産物 | アンドロゲン | 鉱質 コルチコイド |
|---|---|---|---|---|---|
| 不明1 | STAR | ミトコンドリア膜でのコレステロール輸送に関与 | 欠乏2 | 欠乏3 | |
| 不明1 | 3β-HSD | Pregnenolone, 17-OH pregnenolone, DHEA |
Progesterone,17-OHP, Δ4-androstenedione |
欠乏2 | 欠乏3 |
| 不明1 | 17α- hydroxylase |
pregnenolone | 17-OH pregnenolone | 欠乏2 | 過剰4 |
| progesterone | 17-OH (17-OHP) | ||||
| >90% | 21-hydroxylase | progesterone | Deoxycorticosterone (DOC) | 過剰5 | 欠乏3 |
| 17-OH progesterone | 11-deoxycortisol | ||||
| 5% | 11β- hydroxylase |
deoxycorticosterone | corticosterone | 過剰5 | 過剰4 |
- 疾患数が少ないため、不明
- 男性では出生時男性化はない.
- 塩類喪失を伴う.
- 高血圧を伴う.
- 女性では出生時もしくはその後男性化を示す.
非古典型 21-OHD CAH アンドロジェン過剰による症状を示す女性では非古典型 21-OHDを考慮する必要がある.アンドロジェン過剰を伴う女性における非古典型21-OHDの頻度は一般人口の1-3%と報告されているが,人口集団によってはそのような女性の頻度はもっと高い.
チトクロームP450酸化還元酵素欠損症. CAHのまれな病型でTable5には含まれていない.CYP450酸化還元酵素欠損症でPORの変異による.P450C17(17-ハイドロキシラーゼ)およびP450C21(21-ハイドロキシラーゼ)の2つのステロイド合成酵素の部分欠損が同時に存在することが尿中ステロイド排泄から示唆される.注目すべきは,CYP450酸化還元酵素がNADPHからの両酵素への電子伝達に重要な役割を持っているということである.
CYP450酸化還元酵素欠損症の表現型は単一のステロイド異常症から古典型Antley-Bixler症候群(ABS)まで幅広い.POR欠損者はコルチゾールが欠如しており臨床的にたいしたことのないものから生命の危機に及ぶものまである.新生児男児は外性器形態があいまいで,陰茎は小さく停留精巣を認める.新生児女児は膣閉鎖症を認め,小陰唇の融合,大陰唇の低形成,陰核肥大を呈する.ABSの頭蓋顔面の特徴は,POR領域での最も重症例では頭蓋骨癒合症,後鼻孔狭窄または閉鎖症,外耳道狭窄,水頭症を起こすことがある.骨奇形では橈骨上腕骨癒合,新生児期骨折,先天性長幹骨湾曲,屈指症,関節性拘縮,クモ指症,内反足を起こすことがある.遺伝形式は常染色体劣性遺伝である.
臨床的マネジメント
病気の程度を確定するための初期診断における評価
疾患の程度や21-OHD CAHと診断された個人の必要を確立するために、次の評価が勧められる
塩類喪失型を評価するために
- 血清レニン活性(PRA)
- 血清電解質
21-OHD CAHの古典型と非古典型を区別するために
- 17-OHP,Δ4-アンドロステンジオン,コルチゾール,アルドステロンの基礎値
- ACTH刺激試験を実施し,基礎値と負荷後の17-OHPの濃度の比較
女児において出生前の男性化の程度評価のために:
- 外性器形態とその開口についての身体所見の注意深い観察
- 尿道と膣の解剖学的評価のため膣造影
男児,女児ともに出生後の男性化の程度評価のために
- 骨年齢による骨成熟の程度
- 血清の副腎アンドロゲン濃度(DHEA,Δ4-アンドロステンジオン,テストステロン)
21-OHD CAHの診断がついた新規患者は,遺伝カウンセラーと臨床医へのコンサルトが勧められる.
病気の徴候に対する治療
21-OHD CAHの患者の治療のための臨床ガイドラインは出版されている[Speicer et al 2010].
臨床医は早期に治療を開始し,もしあるのならばコルチゾール欠乏や鉱質コルチコイド欠乏による影響を抑えるために, 21-OHDの診断をできる限り早期につける義務がある.
小児内分泌,小児泌尿器,臨床遺伝学,精神科などのさまざまな専門分野のチームがあいまいな外性器の児の診断や治療に必要となる[Hughesら 2006]. 従来のCAH治療センターの草分けのプロジェクトで補完されている[Auchus et al 2010]. 様々な科のケアをすることができる2つの従来のCAHケアセンターは、成長・発達の全時期を通じて、様々な科のケアをすることができるようアメリカでは企画されている.
古典型21-OHD CAH
グルココルチコイド補充療法. グルココルチコイド補充療法の目標は,欠損しているステロイドの補充をおこない,副腎性ホルモンとグルココルチコイド過剰分泌をできる限り少なくすることであり,男性化を予防し成長の可能性を最大限に伸ばし,妊孕性を促進することである[Claytonら 2002].
- ハイドロコルチゾン錠は成長している子供の治療薬である.CAHの治療では基本的にグルココルチコイド補充が必要となり,通常 1日2ないし3回に分けてハイドロコーチゾン(24時間あたり10-20mg/m 2)が経口的に投与される[New et al 2013].小児におけるグルココルチコイド療法は,医原性クッシング症候群を起こさないように,そして正常な成長および骨成熟がおこるように副腎アンドロジェン分泌の抑制をするというバランスが求められる.
注:過剰な治療はクッシング様徴候を招くので避けなければならない.しばしば血清17-OHP濃度が生理学的範囲に低下した場合によく起きる.したがって,治療中の患者における17-OHPの正常範囲は,アンドロジェンが性別や性的発達段階に応じた適切なレベルに維持されているという条件で,通常よりも高めに設定(100-1,000 ng/dl)すべきである.
- ストレス状態下(手術,発熱性疾患,ショック)ではすべての 21-OHP患者はより多量のグルココルチコイドを必要とする.典型的には通常の2-3倍量が経口的または経静脈的に投与される.
- 罹患患者は緊急時のステロイド投与量に関する医療情報を携帯するべきである.
- 古典型21-OHD CAHの患者は生涯にわたってグルココルチコイド投与が必要となる.成長が完了した後はより力価の高いグルココルチコイド(プレドニゾロン,デキサメサゾンのような)を用いることができる.これらは小児では成長を抑制する傾向がある.
鉱質コルチコイド補充療法. 塩類喪失型の 21-OHD患者では,9 α-フルドロヒドロコルチゾン(フロリネフ🄬)(0.05-0.2 mg/日 経口)と塩化ナトリウム(1-2g/日を処方や食事に加える)が必要となる.
- 古典型の患者は全員新生児期および乳幼児期は9 α-フルドロヒドロコルチゾンと塩化ナトリウムサプリメントの補充療法をすべきである[Speiser et al 2010].
- 塩化ナトリウムの補充は幼少期以降は必要としないかもしれない.同様に一日に必要とされる鉱質コルチコイドの量も年齢とともに減少してくる.
女性型の性器形成術. 2006のLWPES/ESPE合同コンセンサス参照[Lee et al 2006](full text).
"重篤な男性化(Prader Ⅲ‐Ⅳ)は手術を検討するべきである.適切な時期に,泌尿生殖洞の修復が行われる.なぜなら,不要な勃起組織を除去し,勃起機能や陰核の神経支配を保存するための解剖学的な手順で手術は行われる.完璧な外観よりも機能的な予後が重要視される.一般的に生下1年で行われる美観のための手術は親の苦悩を軽減し,親子の愛情をより強固にする.ただ,この体系的なエビデンスはまだない."
内分泌学会の臨床ガイドライン[Speiser et al 2010] による
"陰核と会陰形成術は幼児のうちに行われるべきであり,経験ある小児内分泌専門医,心理カウンセラー,ソーシャルワーカーと経験を積んだ外科医が行うべきである."
- 最も手術に適した年齢や手術法を検討したランダム比較試験はないが,神経血管温存―陰核形成術や全もしくは部分的泌尿生殖器手術(urogenital mobilization)による膣形成術が勧められる.
- 膣形成術が必要な場合は,手術は思春期後期に行われる.これは膣開口を保つために膣拡張が必要とされるためである.
思春期早発. 21-OHD CAHにおこる思春期早発は黄体ホルモン放出ホルモン (LHRH)誘導体による治療が行われる.
精巣副腎残存腫瘍(TART). グルココルチコイド治療強化によってTARTはサイズを減らし,精巣機能を改善する[Bachelot et al 2008].精巣温存手術(Testis-sparing surgery)は薬物治療に抵抗性の患者に検討されるが,その予後は好ましいものではなく,おそらく管の長期閉塞によるものであろう[Claahsen-van der Grinten et al 2008].生殖補助医療(ART)は,妊孕性のために検討されるかもしれない[Sugino et al.2006].
思春期から大人への移行. 21-OHD CAH に対する治療の改良によって良好な予後および正常と変わらない平均寿命が得られる.しかし,イギリスでの21-OHD CAHの成人の前向き横断研究は次のようなことを示した[Arlt et al 2010].
- 罹患者は著明な低身長や高度BMIを示す.
- 古典的CAHの女性は拡張期血圧が上昇する.
- 研究の参加者では、代謝異常が良く認められた.肥満(41%),高コレステロール血症(46%),インスリン抵抗性(29%),骨塩低下(40%),骨粗鬆症(7%).主観的な健康状態は著明に障害されており,妊孕性も弱くなっていた.
小児期から成人医療への移行は,通常の生活で良い健康状態と生活の質を達成するように,最適な生涯治療をするための重要なステップである[Reisch et al 2011].
- CAHの成人の治療は多くの専門的アプローチを必要とし、その中には専門家による精神的サポートも含まれる[Ogilvie et al 2006a].
副腎摘出術. ホルモン補充療法でコントロールが困難である,もしくはnull変異のホモ接合体の重度21-OHD CAH患者に対する両側副腎摘出術が報告されている[Van Wyk et al 1996,Meyers&Gru 2000].こうした患者はアジソン病患者に対する治療のように,より上手に治療できるかもしれない.しかし,術後の投薬計画のコンプライアンスは非常に重要である.このように,両側副腎摘出術は内服治療がうまくいかない限られた人のみ検討される.手術の前に,コンプライアンスがない場合のリスクを検討すべきである[Speiser et al 2010].
小数だが,副腎摘出術を受けた人の報告があり[Bachelot et al 2008参照],最大で5人の方が含まれていた[Ogilvie et al 2006b].3つの指標がある.不妊,男性化,肥満である.この3つすべての改善が、報告された人すべてで述べられている.副腎摘出術を受ける人の予後を決定するにはより長期のデータが必要である.潜在的ACTH増加は副腎残存組織を悪化させるかもしれないからである.
非古典型21-OHD CAH
非古典型21-OHD患者は必ずしも治療を必要としない.多くは生涯にわたって無症状であり,あるいは症状が思春期,思春期後,あるいは閉経後に現れる.
- 治療を必要とするような高アンドロゲン血症は,次のような状態である.少年期:骨年齢促進,早期の恥毛,早発思春期,高身長,早期骨端閉鎖;成人男性女性両者:不妊,ざ瘡,低身長;女性:男性型多毛,前額禿頭,多嚢胞卵巣,月経不順;男性:精巣副腎残存組織.[New 2006]
- 以前より治療していた方では,症状が寛解したとき,治療中止のオプションを提供すべきである[Speiser et al 2010].
伝統的に,非古典型21-OHD CAHは古典型より低用量のグルココルチコイドによる治療が行われてきた.
初期症状の予防
塩類喪失クリーゼ. 新生児スクリーニングプログラムは古典型21-OHDの新生児を同定し,生命を脅かす塩類喪失クリーゼの可能性がある児に対して事前に初期治療を行う目的がある.
治療と症候を参照.(グルココルチコイド補充療法,ミネラルコルチコイド補充療法)
二次的合併症の予防
低身長. 低身長はグルココルチコイドの過剰治療による成長抑制あるいは不適切なグルココルチコイド治療による骨成熟の促進によるかもしれない.観察研究によるエビデンスではCAHでグルココルチコイド治療していた人の最終的な身長は,一般人口や親から予測される身長よりも低かった[Muthusamy et al 2010].CAHの患者の低身長の治療の議論の調査下治療(Therapies Under Investigation)を参照.
定期検査
以下の評価は子供たちが活発に成長するにあたり3-4カ月ごとに行われるべきである.それ以降はより間隔をあけて行われる.評価の頻度は個々の患者の必要性に応じて変更するべきである[Speiser et al 2010].
グルココルチコイド補充療法の効果は以下の測定でモニターする:
- 早朝血清中の17-OHP,Δ4-アンドロステンジオン,テストステロンの濃度を新生児期はだいたい3ヶ月毎に,その後は3-6ヶ月毎に測定.(いくつかの例では,24時間尿中サンプルを用いて尿中のプレグナントリオール,17-KSがホルモンコントロールの評価に役立つことがある.しかし,蓄尿法は単純採血に比べて実用的ではない.)
- 身長の伸び,体重増加,思春期の発達,コルチゾールやアンドロゲン過剰の臨床徴候.
- 骨成熟度を評価するために骨年齢(6-12カ月間隔で)
ミネラルコルチコイド補充療法の効果は以下の測定でモニターする:
- 血圧
- 早朝血清レニン活性または直接レニン分析(たいていは直立姿勢で)
男性における精巣異常のモニタリング. 超音波やMRIを用いて精巣の定期的な画像診断を思春期以降より開始,3-5年ごとに繰り返し行われるべきである.
成人における妊孕性と代謝異常リスクのモニタリング. 罹患した成人では,次のようなモニタリングや定期的な検査を行う
- 生殖能力か妊孕性
- 体重
- 脂質プロファイル
- 血圧
- 骨塩濃度
画像検査. 定期的な副腎画像検査や骨塩濃度定量は推奨されない[Speiser et al 2010].
避けるべき物質/環境
発熱疾患,脱水を伴う胃腸炎,一般的な麻酔を伴う手術,大きな外相のような身体ストレスは,古典的CAHの患者に副腎クリーゼを引き起こすことがある.このような状況の時は、グルココルチコイドの容量を増やすことが推奨される.
リスクのある血縁者の検査
もし21-OHD CAHの出生前診断が行われていなかった場合,早期診断と治療を容易にするために発端者の新生児の兄弟を評価することは適切である.
- 血清17-OHP濃度は新生児スクリーニングに加えて調べるべきである.
- もし,家族の病的変異がわかっていれば,分子遺伝学的検査は勧められる.
遺伝カウンセリングの項目を参照.
妊娠時のマネジメント
古典的21-OHD CAHの妊娠女性.古典的塩類喪失型の21-OHD CAHの妊娠女性は妊娠中は内分泌医の頻回なモニターを必要とする.妊娠中は副腎アンドロゲンが増加するため、グルココルチコイドの維持量は通常増やす必要がある.妊娠中の副腎アンドロゲンの産生増加にも関わらず、女性胎児の性器は男性化しない[Lo et al 1999].
検査後の治療
あいまいな性器をもった女性罹患胎児. 胎児のDNAの分子遺伝学的検査を通じて、子宮内で21-OHD CAH合成欠損は診断される.女性罹患胎児の性器のあいまいさは妊娠期間早期から出産まで母親のデキサメサゾン投与を続けることによって胎児アンドロゲンの生産が抑制されることによって、軽減または消失するかもしれない.出生前治療は実験的と考えられているので、正式なIRB承認臨床試験の手続きでのみ行える.罹患女性胎児のより早い診断のための非侵襲的出生前診断法が開発され、男児や罹患女児の不要な出生前治療がなくなるかもしれない[New et al 2014, Tardy-Guidollet et al 2014].
低身長の治療. ヒト型成長ホルモン単独もしくはゴナドトロピン放出ホルモン(GnRH)との併用療法は,著明な成長障害を起こした21-OHD CAH罹患者の身長を改善するために使用される[Lin-Su et al 2011].骨年齢の成長を緩やかにするためにアロマターゼ抑制薬が使われる.しかし,これらのアプローチは経験的治療と考えるため,正式な臨床試験外では使用されるべきではない[Speiser et al]. ClinicalTrials.gov参照.
注:この疾患のための臨床試験はないかもしれない.
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
先天性副腎過形成 21水酸化酵素欠損症(21-OHD CAH)は常染色体劣性遺伝である.
患者家族のリスク
発端者の両親
- ほとんどの両親はヘテロ接合体である(すなわち、1アレルのCYP21A2病的変異の保因者である).
- ヘテロ接合体は ACTH負荷を行ったときの17-OHPレベルが正常アレルのみを持つ人に比べてわずかに上昇していることがあるものの,無症状である(Carrier Detectionを参照).
- 約1%のCYP21A2変異は新生突然変異で生じるので,発端者の1%では両親のうち一人だけがヘテロ接合体である[Kroneら2000].
- 時に罹患しているとは思われていなかった親が非古典型 21-OHDであることが判明することがある.両親が非古典型21-OHDを有していないかどうか明らかにするために,分子遺伝学的検査やホルモン検査を施行するのは適切である.
発端者の同胞
- し患者の両親がヘテロ接合体であるならば,個々の同胞について,変異アレルを 2つ受け継ぎ,その結果罹患する確率は25%,変異アレルを1つだけ受け継いだ無症候保因者である確率が50%,正常アレルのみを受け継いだ非保因者である確率が25%である.
- もし患者の片親がヘテロ接合体でもう片方の親が21-OHD CAH発症者であるならば,個々の同胞について,変異アレルを 2つ受け継ぎその結果罹患する確率は50%,変異アレルを1つだけ受け継いだ無症候保因者である確率が50%である.
- ヘテロ接合体をもつ保因者は無症候性であるが,2つとも野生型アリルをもつ人に比べると,ACTH負荷にて軽度17-OHPが上昇することがある(Carrier Detection参照).
発端者の子
- 罹患した患者は子供に変異アレルを 1つ伝える.
- 21-OHDの保因者頻度が高いので,患者のパートナーに対して CYP21A1 の分子遺伝学的検査を提供するのは適切である.
- もしパートナーが非保因者であれば,子が21-OHDに罹患する可能性はずっと低くなる.
- もしパートナーが既知の変異の保因者であった場合,二人の子が罹患する可能性は50%である.遺伝子型にもとづいて臨床型を予測するのは不完全にしかできない. (Genotype-Phenotype Correlations参照)
発端者の他の血縁者
発端者に変異を伝えた親の同胞も 50 %の確率で保因者である.
保因者検査
分子遺伝子学的検査.
CYP21A2 遺伝子の分子遺伝学的検査によるリスクのある親族の保因者検査は,家族に原因となる変異が同定されていることが必要である.
注:可能性のある誤診の別の原因としてCYP21A2重複がある[Koppens et al 2002].ある研究では、調べた人口のCYP21A2アリルの7%に重複があった[Parajes et al 2008].よくある重複のハプロタイプは,同じ染色体上に野生型CYP21A2アリルと一緒にp.Gln319Terの病的変異アリルが存在するものである[Kleinle et al 2009].これは、保因者でない人の保因者スクリーニングで偽陽性の結果となりうる.同じ染色体上に機能的な遺伝子と病的変異をもつ重複コピー遺伝子をもつ人は,正しくないことだが保因者のラベルをされるかもしれない.これは、誤った出生前診断を誘導するかもしれない[Lekarev et al 2013].このような人々は欠損/重複解析かハプロタイプ解析によって同定されうる.
ホルモン検査
保因者では非保因者に比べて ACTH負荷時の17-OHPレベルがわずかに高くなるが,これは保因者と非保因者との間でオーバーラップがある.したがって,保因者検査には分子遺伝学的検査が好ましい.
遺伝カウンセリングに関連した問題
早期診断、早期治療の目的でのリスクのある血縁者の情報のためのリスクのある血縁者の評価,マネジメントの項参照.
家族計画
- 遺伝学的リスクの確定,保因者の診断,出生前診断の利用の議論の最適な時間は妊娠前である.
- 罹患した,または保因者のリスクがある若い成人に(子供の潜在的なリスク子を伝え、子供を作る選択の議論を含む)遺伝カウンセリングを提供するのは適切である.
DNAバンク DNAバンクとは将来的な使用を想定してDNAを(一般的には白血球から抽出する)保存しておくものである.遺伝子検査技術や遺伝子,変異,疾患に対するわれわれの認識が将来変化するかもしれないので,現在の遺伝子検査の精度がすべての変異を検出できないような場合にはDNA保存が考慮されうる.
出生前診断と着床前診断
高リスク妊娠. 一度罹患家族にCYP21A2病的変異が認められれば,妊娠時にリスクと着床前診断のための21-OHD CAHの遺伝学的診断は可能である.
罹患女児の胎児の非侵襲的早期診断のための出生前診断法は開発しており,出生前の男性と非罹患女児の不要な治療をなくせるかもしれない[New et al 2014, Tardy-Guidollet et al 2014].
低リスク妊娠. 出生前の超音波検査の使用頻度の増加や解像度の改善によって胎児期の生殖器異常や副腎異常がより高頻度に同定されるようになっている[Saada et al 2004].Pinhas-Hamielらは2002年に,出生前に超音波検査をされた10000人の胎児のうち16人で性器形態異常を同定した.16人中3人は最終的に21-OHD CAHの診断であった.
もし定期超音波検査で外性器形態があいまいであった場合には,胎児の染色体型,SRY遺伝子に対するFISH,そしてミュラー管構造物の超音波による評価が必要である.46XX,SRY(-),子宮形態が正常の胎児は古典型21-OHDを考慮しなくてはいけない.羊水穿刺もしくは胎盤絨毛採取にてCYP21A2の分子遺伝子学検査が望ましい. 生化学出生前診断は研究されているが,この疾患の診断には適切ではない.
低リスク妊娠での21‐OHD CAH罹患胎児の診断は出生前治療をするためにするわけではないが,21-OHD CAHの出生前診断は新生児期のマネジメントや家族の準備の面で意味のあることである.
資料
GeneReviewsスタッフは罹患者と家族のための疾患特異的もしくは支持組織もしくは登記を選別した.GeneReviewsは他団体による情報について責任を負わない。
- CARES foundation
- Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency: A guide for patients and their families
- National Library of Medicine Genetics Home Reference
- NCBI Genes and disease
- Save Babies Through Screening Foundation, Inc.
分子遺伝学
分子遺伝学
Molecular geneticsやOMIMの情報と,このGeneReviewの項目に記載されているものは違うかもしれない.表には,より最近の情報が含まれている可能性がある.
Table A. 21-OHD CAH: Genes とDatabases
| 遺伝子 | 染色体座 | 蛋白 | Locus Specific | HGMD |
|---|---|---|---|---|
| CYP21A2 | 6p21?.33 | Steroid 21-hydroxylase | CYP21A2 @ Human Cytochrome P450 (CYP) Allele Nomenclature Committee CYP21A2 database |
CYP21A2 |
Table B. 21-OHD CAH: OMIM Entries (OMIM参照)
| 201910 | ADRENAL HYPERPLASIA, CONGENITAL, DUE TO 21-HYDROXYLASE DEFICIENCY |
| 613815 | CYTOCHROME P450, FAMILY 21, SUBFAMILY A, POLYPEPTIDE 2; CYP21A2 |
遺伝子構成. 副腎の21水酸化酵素の機能性遺伝子,CYP21A2は非機能性の偽遺伝子CYP21A1Pから30kbほどの場所にありHLA遺伝子クラスターの6p染色体上に存在する.
CYP21A2遺伝子と多数の病的変異をもつため不活性化されているCYP21A2P遺伝子は高レベルのヌクレオチドシークエンス相同性がある(エクソンの98%,イントロンの96%).両者は10個のエクソンから構成される.詳細は、Table A Gene参照.
良性変異. 機能的CYP21A2遺伝子の5つの良性変異はTable 6参照.
病的変異. CYP21A2とCYP21A1P には繰り返す(二重)ように縦に配列している場所がある.この配列は繰り返しシークエンスの間で組み換えを促進させる.その組み換えは21-OHD CAHを起こすCYP21A2の病的変異の主な原因となる.機能的なCYP21A2相同体間での減数分裂中の不均等な交差の組み換えの結果,大きなCYP21A2の欠失や重複が起こる.高度のCYP21A2とCYP21A2P間では非常にシークエンスが似ているため、遺伝子変換が促進され[Higashi et al 1988, Tusie-Luna & White 1995, Wedell 1998],それによって機能するCYP21A2遺伝子がCYP21A1P偽遺伝子の部分的コピーに置き換わる現象が起こる.それゆえ,変換したCYP21A2の部分は偽遺伝子の典型的な変異シークエンスになっている.このような変異は病的であり、機能的なCYP21A2遺伝子発現と機能的な蛋白翻訳を不活化させる
- 小範囲の遺伝子変換は,アリル特異的PCR法で検出される,同じアリル上にあるP.Pro31Leu, c.293-13AorC>G, p.Gly111ValfsTer21など同じ病的変異の存在を説明する.
- 広範囲の遺伝子変換も起こり,そのいくつかは,追加検査が必要となる(確立された診断,分子遺伝学の項目参照).
- 変異アリルの約20-30%は減数分裂中の繰り返し配列の組み換えの結果であり,CYP21A1P偽遺伝子の3末端,隣接するC4B補体遺伝子の全て,CYP21A2遺伝子の5末端を含む30kbの遺伝子欠損を起こし,それゆえ機能の失われたキメラ偽遺伝子が生じる[White et al 1988].
- 別のよくある病的変異は,c.293-13A>Gまたはc.293-13C>Gであり,20-30%の頻度で起きる.それは,異常なスプライシングを起こし,端の短いまたは異常な蛋白ができる.
機能の失われた偽遺伝子の9つの病的変異は,遺伝子変換によってCYP21A1P遺伝子からCYP21A2遺伝子へ乗り換えを起こしたときに,機能のある遺伝子を不活化する[Wedell 1998].CYP21A2欠失や明らかに広範囲の遺伝子転換とともに,これらの9つの病的変異はすべての疾患原因CYP21アリルの約95%を占める[Wedell 1998].
1塩基多型,小欠失,小塩基挿入,遺伝子再構成を含む100以上の病的変異が報告されている.
(より詳細は,Table A参照)
表 6. CYP21A2変異
| 変異分類 | DNA ヌクレオチド変化 (別表記 1) |
予測される蛋白変異 (別表記 1) |
参考とするシークエンス |
|---|---|---|---|
| 良性 | c.25_27dupCTG | p.Leu9dup2 | NM_000500.5 NP_000491.2 |
| c.308G>A | p.Arg103Lys (Lys102Arg) |
||
| c.552C>G | p.Asp184Glu (Asp183Glu) |
||
| c.806G>C | p.Ser269Thr (Ser268Thr) |
||
| c.1482C>T | p.Asn494Ser (Asn493Ser) |
||
| 病的 | c.92C>T | p.Pro31Leu (Pro30Leu) |
|
| c.293-13A>G (659A>G) |
-- | ||
| c.293-13C>G (659C>G) |
-- | ||
| c.332_339del (8-bp deletion in exon 3 or 707_714del) |
p.Gly111ValfsTer21 (G110_Y112delfs) |
||
| c.518T>A | p.Ile173Asn (Ile172Asn) |
||
| c.[701T>A;713T>A;719T>A] | p.[Ile237Asn;Val238Glu;Met240Lys] (I236N, V237E, M239K) (exon 6 mutation cluster) |
||
| c.844G>T | p.Val282Leu (Val281Leu) |
||
| c.844G>C | p.Val282Leu (Val281Leu) |
||
| c.923dupT (Leu307insT) |
p.Leu308PhefsTer6 (F306+T) |
||
| c.955C>T | p.Gln319Ter (Gln318Ter) |
||
| c.1069C>T | p.Arg357Trp (Arg356Trp) |
||
| c.1360C>T | p.Pro454Ser (Pro453Ser) |
||
| Entire gene deletion | -- | ||
| Entire gene duplication | -- |
変異の分類に関して注意:テーブルの変異リストは著者によるもので,GeneReviewsスタッフは独自に確認はしていない.
命名法による注意:GeneReviewsはHGVS(Human Genome Variation Society)による標準的な命名法に従っている.命名法説明のQuick Reference参照.
- 変異の現在の表記は固定ではない
- Higashi et al [1986], White et al [1986]
正常遺伝子産物. コードされている蛋白は494のアミノ酸からなり分子量55kdの蛋白である.この酵素は多くとも28%のその他のチトクロームP450酵素と同種である.
異常遺伝子産物. 異常産物は特異的変異に依存する.およそ20%の変異は30Kbの欠損である.病的変異の約20%は減数分裂時の遺伝子組み換えによる30kbの遺伝子の欠失であり、それはCYP21A1P偽遺伝子の3末端,隣接するC4B補体遺伝子の全て,CYP21A2遺伝子の5末端を含み,無機能のキメラ偽遺伝子を生じる.更新履歴
- Gene Review著者:Maria I New, MD ; Saroj Nimkarn, MD
日本語訳者: 塚本幸子,臼井健 (国立病院機構京都医療センター)
Gene Review 最終更新日: 2006.9.7. 本語訳最終更新日: 2007.9.21 - Gene Reviews著者: Saroj Nimkarn, MD, Prasanna K Gangishetti, MBBS, Mabel Yau, MD, and Maria I New, MD.
日本語訳者:和泉 賢一 (札幌医科大学医学部遺伝医学,NGSDプロジェクト)
Gene Reviews 最終更新日: 2016.2.4 日本語訳最終更新日: 2017.12.29 (in present)
原文 21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia
| X POST |
![]()