








Guidelines on Hereditary Leukodystrophies
CQ 3-3 Hypomyelination with atrophy of the basal ganglia and cerebellum (HABC; HLD6, OMIM #612438)
Hypomyelination with atrophy of the basal ganglia and cerebellum (HABC) is a form of hypomyelination of the central nervous system caused by a heterozygous mutation of the TUBB4A gene. HABC occurs sporadically due to a de novo mutation. In addition to hypomyelination being visible on magnetic resonance imaging, atrophy of the basal ganglia and cerebellum are evident, and these imaging findings give the disease its name. Delayed motor development is followed by regression, extrapyramidal tract symptoms (dystonia), motor ataxia, and spastic palsy. Intellectual disability is milder than motor disability. Patients with the high-frequency mutation c.745G>A have milder symptoms than those with other mutations. At this point, symptomatic therapy is provided.
Disease concept
In hypomyelination with atrophy of the basal ganglia and cerebellum (HABC), in addition to congenital white-matter hypomyelination, progressive atrophy of the basal ganglia (most obviously in the caudate nucleus and putamen) and the cerebellum are evident. It is these characteristic imaging findings that give the disease its name. HABC was first reported by van der Knaap et al. in 2002, and the disease concept was established in 2007.1,2) In 2013, a TUBB4A mutation was shown to cause HABC.3) TUBB4A has also been reported as the causative gene of dystonia 4 (c.4C>G [R2G]).4) All 11 of the patients described in the initial report carried the high-frequency mutation c.745G>A (D249N),3) but other mutations have subsequently been reported.5-8) Almost all cases are sporadic, and the de novo mutation exhibits autosomal dominance.
Epidemiology
The incidence is unknown, with 71 cases reported worldwide to date.9)
Etiology and pathophysiology
The heterodimer formed by b-tubulin and a-tubulin plays an important role in the formation of microtubules, an essential part of the cytoskeleton. The brain-specific b-tubulin encoded by TUBB4A (tubulin b-4A) is expressed at high levels in the cerebellum, putamen, and white matter (particularly oligodendrocytes).4) In the central nervous system, tubulin is important for neuronal formation and maintenance. Most reported TUBB4A mutations are located at the a-b-tubulin junction.3, 5-8) These mutations prevent the formation or maintenance of heterodimers, causing microtubule dysfunction of the neurons and oligodendrocytes that results in secondary glial damage3) and damages myelination.
Clinical symptoms
Most patients do not exhibit any abnormalities at birth, including nystagmus. The timing and severity of the appearance of motor and intellectual disabilities, pyramidal tract symptoms (spastic palsy), extrapyramidal tract symptoms (such as dystonia, chorea, athetosis, and rigidity), cerebellar symptoms (such as ataxia), and bulbar symptoms (such as dysarthria and dysphagia) vary widely. Typical patients carrying the high-frequency mutation c.745G>A (25 cases) have milder symptoms than those with other mutations (16 cases).6) The maximum attainable motor function is walking unaided, achieved by 76% of patients with the TUBB4A c.745G>A mutation, but of those with other mutations, 0% were able to walk unaided, 12% could walk with assistance, 38% could maintain a seated position, 25% could extend their arm and grasp objects, and 25% could perform voluntary movements. In terms of maximum attainable language function, normal speech was achieved by 38% of patients with the c.745G>A mutation vs. 0% of patients with other mutations, individual words or short sentences by 68% vs. 6% and no speech acquisition by 0% vs. 94%. Maximum attainable intellectual level was normal in 32% vs. 6%, low in 68% vs. 38%, and perception only in 0% vs. 56%. Neurological symptoms comprised spasticity in 96% vs. 94%, extrapyramidal symptoms in 96% vs. 100%, seizures in 12% vs. 53%, short stature in 40% vs. 87%, low body weight in 48% vs. 88%, and microcephaly in 9% vs. 69%. Extrapyramidal symptoms occur in almost all HABC patients, a clinical difference with other forms of congenital leukodystrophy.
Investigations
Although very slight hypointensity of the corpus callosum and the posterior limb of the internal capsule may be evident on T2-weighted imaging, almost all of the white matter appears hyperintense. The signal on T1-weighted imaging varies from hypointense to very slightly hyperintense. There is also progressive atrophy of the cerebellum (vermis > hemisphere), caudate nucleus and putamen, and cerebral white matter (Figure 3). The globus pallidus and thalamus are spared. By 2 years after onset, 70% of patients exhibit atrophy of the putamen, 30% of the caudate nucleus, 22% of the cerebrum, 17% of the corpus callosum, and 81% of the cerebellum. Between 2 and 12 years after onset, the corresponding rates are 97%, 53%, 44%, 59%, and 94%, while more than 12 years after onset they have progressed to 100%, 90%, 80%, 100%, and 100%.6) In some patients with a TUBB4A mutation, there is no atrophy of the basal ganglion and only the cerebellum is atrophied, making it difficult to distinguish from HDL7 (POLR3A mutation) and HDL8 (POLR3B mutation) on the basis of imaging findings alone.5,7,8) Some patients may not exhibit atrophy of either the basal ganglia or the cerebellum.8)
Genetic diagnosis
Heterozygous mutations (point mutations) of the TUBB4A gene have been reported. The first 11 reported cases all carried the high-frequency mutation c.745G>A (D249N),3) but there have been numerous reports of other mutations. Most cases are sporadic, but brothers born to a mother with somatic cell mosaicism was reported.3)
Treatment and care
At this point, treatment is limited to symptomatic therapy.9) The use of l-dopa has been reported to improve the symptoms of dystonia, a clinical characteristic of HABC.8)
Figure.
Hypomyelination with atrophy of the basal ganglia and cerebellum.
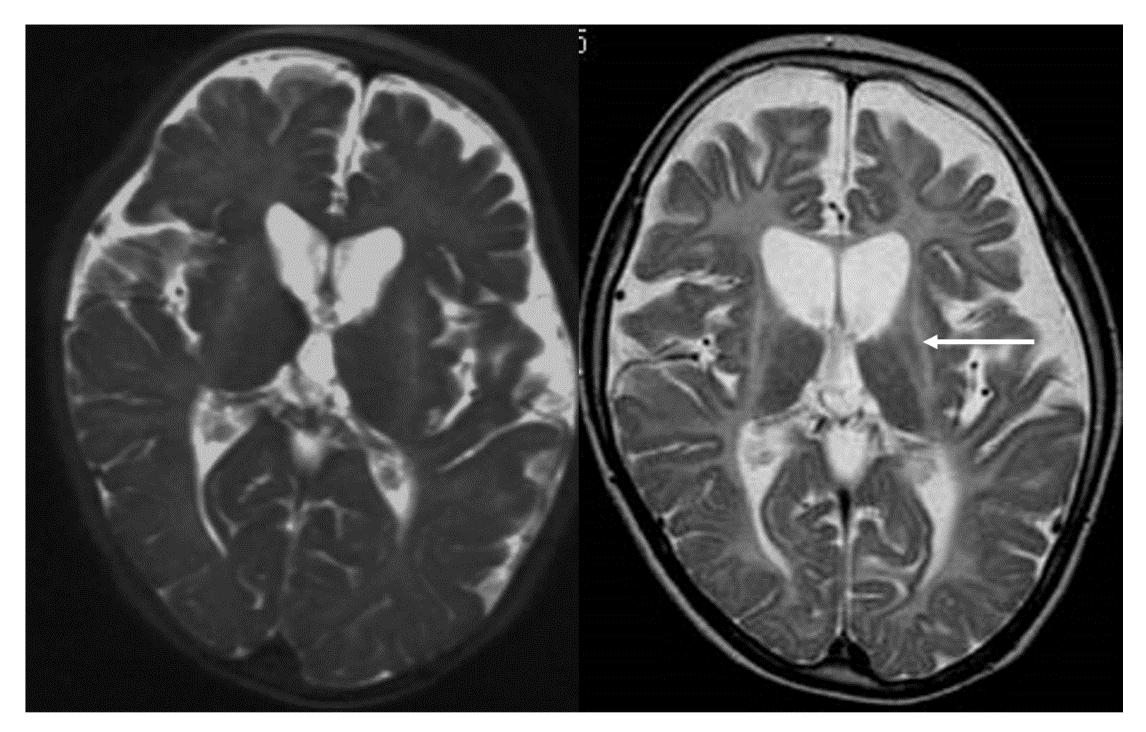
(Photograph kindly provided by Dr. Shinji Fujimoto and Dr. Ayako Hattori of the Department of Pediatrics, Nagoya City University.)
T2-weighted imaging during infancy (left) shows hyperintensity of the white matter and atrophy of the basal ganglia and cerebellum. T2-weighted imaging taken during later childhood (right) clearly shows progression of the atrophy of the basal ganglia (arrow) and cerebellum.
References
(Evidence levels are shown in parentheses.)
- van der Knaap MS, Naidu S, Pouwels PJ, et al. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol 2002; 23: 1466-1474.(5)
- van der Knaap MS, Linnankivi T, Paetau A, et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: follow-up and pathology. Neurology 2007; 69: 166-171.(5)
- Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 2013; 92: 767-773.(5)
- Hersheson J, Mencacci N, Davis M, et al. Mutations in the autoregulatory domain of beta-tubulin 4a cause hereditary dystonia. Ann Neurol 2013; 73: 546-553.(5)
- Miyatake S, Osaka H, Shiina M, et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology 2014; 82: 2230-2237.(5)
- Hamilton EM, Polder E, Vanderver A, et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation. Brain 2014; 137: 1921-1930.(5)
- Pizzino A, Pierson TM, Guo Y, et al. TUBB4A de novo mutations cause isolated hypomyelination. Neurology 2014; 83: 898-902.(5)
- Tonduti D, Aiello C, Renaldo F, et al. TUBB4A-related hypomyelinating leukodystrophy: New insights from a series of 12 patients. Eur J Paediatr Neurol 2016; 20: 323-30.(5)
- Nahhas N, Conant A, Hamilton E, et al. TUBB4A related leukodystrophy. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017.(6)
PubMed search
- TUBB4A[All Fields] AND leukodystrophy[All Fields] 11 results
- H-ABC[All Fields] 44 results