








Guidelines on Hereditary Leukodystrophies
Pelizaeus-Merzbacher-like disease 1 (PMLD1, HLD2, OMIM#311601)
Disease description: Patients with symptoms indistinguishable from those of Pelizaeus-Merzbacher disease (PMD) but who do not exhibit a PLP1 mutation are distinguished by the diagnosis of Pelizaeus-Merzbacher–like disease (PMLD). This is a rare disease. In some patients who exhibit an autosomal recessive pattern of inheritance, it is caused by a mutation in the gap junction protein C2 gene (1), in which case it is known as PMLD1. On magnetic resonance imaging, the pyramidal tract is comparatively spared compared with PMD and motor development is thus comparatively better, with patients becoming able to sit up and walk. However, regression starts by the teen years. There is currently no effective treatment, and only symptomatic care is provided.
1Overview
Definition
Patients with symptoms indistinguishable from those of Pelizaeus-Merzbacher disease (PMD) but who do not exhibit a PLP1 mutation are distinguished with the diagnosis of Pelizaeus-Merzbacher-like disease (PMLD). They include cases formerly reported as female PMD patients. This is a rare disease. In some patients who exhibit an autosomal recessive pattern of inheritance, it is caused by a mutation in the gap junction protein C2(GJC2) gene (1), in which case it is known as PMLD1.
Epidemiology
Unknown
Etiology and pathophysiology
GJC2 (also known as Cx47 or GJA12) mutations were reported in 2004, demonstrating that this condition is genetically different from PMD (1). However, GJC2 mutations account for only 8% of all cases (2), and many PMLD patients do not exhibit GJC2 mutations.
GJC2 encodes the gap junction protein (connexin-47), which plays an important role in myelination of the central and peripheral nerves. There are 20 different types of connexin in mice and 21 in humans, of which C47, C32, and C29 are expressed in oligodendrocytes and C26, C30, and C43 in astrocytes. Coupling between oligodendrocytes and astrocytes is regulated by the expression of C47 and C43 and the expression of C32 and C30, which are essential to the maintenance of myelin (3).
Cx47 mutations cause loss of the gap junction function or prevent hemichannel formation on the oligodendrocyte cell membrane surface (3).
Clinical symptoms
Nystagmus becomes apparent within 6 months of age, cerebellar ataxia by 4 years of age, and spasticity by 6 years of age. Sensory and motor peripheral neuropathy may also be present (4).
In a study of 33 patients, 12 became capable of walking unaided and demonstrated better maximum motor development than that seen in PMD. However, in all cases, regression started before 10 years of age; by age 10, the patients had lost the ability to walk with support. Some mild cases following the same trajectory as spastic paraplegia have also been reported (5).
Imaging and other investigations
Cranial MRI shows diffuse hyperintensity of the white matter on T2-weighted imaging, but the corticopyramidal tract is comparatively spared (2).
Another MRI study of 112 patients with PMD and other forms of cerebral leukodystrophy found that T2 hyperintensities of the pons were only evident in the pyramidal tract or the pons as a whole in 6 of 15 patients with PMLD1; in another 6, the pons was hyperintense on T2-weighted imaging and hypointense on T1-weighted imaging (6).
In physiological testing, waves III–IV of the brainstem auditory-evoked potential are lost in patients with PMD, but one study of 10 PMLD1 patients detected these waves in 11 of 13 tests. Nerve conduction test results are normal in PMD, but a study of PMLD1 found that 2 of 10 patients exhibited signs of mild neuropathy (7)
Magnetic resonance spectroscopy testing of two PMLD1 patients found that levels of choline, N-acetyl aspartate, and creatine were normal or close to normal (2).
Genetic diagnosis
Genetic diagnosis is performed by mutation detection using sequencing or other methods.
2 Treatment and care
Since there is currently no treatment that can cure congenital leukodystrophy, symptomatic care is provided. (See CQ4.)
3 Diet and nutrition
No particular diet or type of nutrition is recommended. A nutritionally balanced diet is desirable.
4 Prognosis
There has been no survey of the natural history of PMLD, but in a study of 33 patients, 12 became capable of walking unaided, better maximum motor development than that seen in PMD. In all cases, regression started before 10 years of age; by age 10, the patients had lost the ability to walk with support. Some mild cases following the same trajectory as spastic paraplegia have also been reported (5).
5 Differential diagnosis
PMLD must be distinguished from PMD. PMD includes more clinically serious cases and a variety of changes, including diffuse white-matter lesions, PLP1 gene duplication, point mutations, and gene deletions. Other non-congenital diseases causing cerebral leukodystrophy must also be included in the differential diagnosis. (See Figure 1.)
6 Recent topics
Figure legends
Figure 1. Differentiation on magnetic resonance imaging
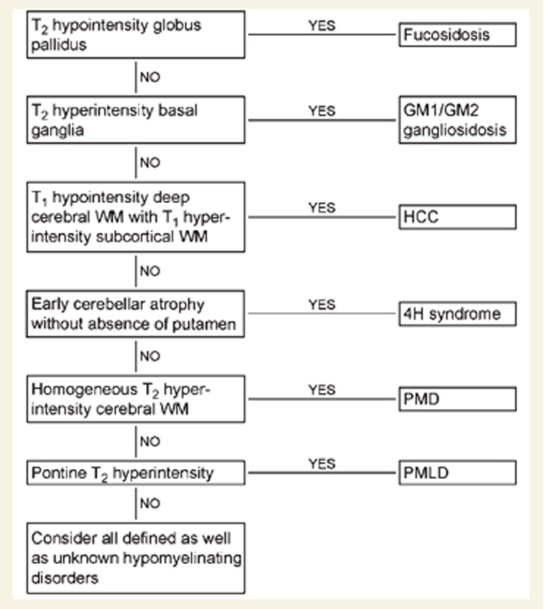
hypomyelination with congenital cataract(HCC)、hypomyelization with hypogonadotropic hypogonadism(4H)
Literature search
PubMed
- pelizaeus-merzbacher-like[All Fields] AND ("disease"[MeSH Terms] OR "disease"[All Fields])
57 results
References
(Unless otherwise noted at the end, all are evidence level 6.)
- Uhlenberg B, Schuelke M, Ruschendorf F, Ruf N, Kaindl AM, Henneke M, et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. American journal of human genetics. 2004 Aug;75(2):251-60. PubMed PMID: 15192806. Pubmed Central PMCID: PMC1216059. Epub 2004/06/12. eng. (Level 5)
- Abrams CK, Scherer SS. Gap junctions in inherited human disorders of the central nervous system. Biochimica et biophysica acta. 2012 Aug;1818(8):2030-47. PubMed PMID: 21871435. Pubmed Central PMCID: PMC3771870. Epub 2011/08/30. eng.
- Diekmann S, Henneke M, Burckhardt BC, Gartner J. Pelizaeus-Merzbacher-like disease is caused not only by a loss of connexin47 function but also by a hemichannel dysfunction. European journal of human genetics : EJHG. 2010 Sep;18(9):985-92. PubMed PMID: 20442743. Pubmed Central PMCID: PMC2987409. Epub 2010/05/06. eng.
- Karimzadeh P, Ahmadabadi F, Aryani O, Houshmand M, Khatami A. New mutation of pelizaeus--merzbacher-like disease; a report from iran. Iranian journal of radiology : a quarterly journal published by the Iranian Radiological Society. 2014 May;11(2):e6913. PubMed PMID: 25035705. Pubmed Central PMCID: PMC4090646. Epub 2014/07/19. eng. (Level 5)
- Osaka H, Hamanoue H, Yamamoto R, Nezu A, Sasaki M, Saitsu H, et al. Disrupted SOX10 regulation of GJC2 transcription causes Pelizaeus-Merzbacher-like disease. Annals of neurology. 2010 Aug;68(2):250-4. PubMed PMID: 20695017. Epub 2010/08/10. eng. (Level 5)
- Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, Mancini GM, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain : a journal of neurology. 2010 Oct;133(10):2971-82. PubMed PMID: 20881161. Pubmed Central PMCID: PMC3589901. Epub 2010/10/01. eng. (Level 3)
- Henneke M, Gegner S, Hahn A, Plecko-Startinig B, Weschke B, Gartner J, et al. Clinical neurophysiology in GJA12-related hypomyelination vs Pelizaeus-Merzbacher disease. Neurology. 2010 Jun 01;74(22):1785-9. PubMed PMID: 20513814. Epub 2010/06/02. eng. (Level 3)