チロシン血症Ⅰ型
(Tyrosinemia Type Ⅰ)
[同義語: FAH Deficiency, Hepatorenal Tyrosinemia, Hereditary Tyrosinemia Type Ⅰ]
Gene Reviews著者: Lisa Sniderman King, MSc, CGC, Crisine Trahms, MS, RD, and C Ronald Scott, MD.
日本語訳者: 和田宏来 (県西総合病院小児科/筑波大学大学院小児科)
Gene Reviews 最終更新日: 2017.5.25.日本語訳最終更新日: 2017.7.24 (minor revision: 2017.10.30)
要約
疾患の特徴
未治療のチロシン血症Ⅰ型では、通常は乳児期早期に重度の肝障害を認めるか、乳児期後期に成長障害やくる病を合併する肝機能障害および腎尿細管機能障害を認める。未治療の小児患者では、反復性でしばしば気付かれない神経性クリーゼ(neurologic crises)が1-7日間持続する可能性がある。神経性クリーゼでは、精神状態の変化、腹痛、末梢神経障害、人工呼吸を必要とする呼吸不全をきたしうる。未治療患者は通常10歳以前に死亡するが、典型的には肝不全、神経性クリーゼ、肝細胞癌による。ニチシノンと低チロシン食の併用療法により、90%を超える生存率、正常な成長、肝機能の改善、肝硬変の予防、尿細管性アシドーシス、および二次性くる病の改善が認められる
診断・検査
チロシン血症Ⅰ型はFAH遺伝子にコードされるフマリルアセト酢酸ヒドラーゼ(fumarylacetoacetate hydrolase, FAH)の欠損による。典型的な生化学的所見(血中および尿中サクシニルアセトン濃度の上昇、血漿チロシン・メチオニン・フェニルアラニン濃度の上昇、尿中のチロシン代謝物およびδアミノレブリン酸化合物濃度の上昇)および/または分子遺伝学的検査でFAH遺伝子両アレル病原性変異を同定することによって診断される。
臨床的マネジメント
症候の治療:
ニチシノン(オーファディン, Orfadin®)、2-(2-ニトロ-4-トリフロロ-メチルベンゾイル)-1, 3 シクロヘキサンジオン(NTBC)はチロシン分解経路の第2段階であるパラヒドロキシフェニルピルビン酸ジオキシゲナーゼ(parahydroxyphenylpyruvic acid dioxygenase, p-HPPD)を阻害し、フマリルアセト酢酸の蓄積とそのサクシニルアセトンへの変換を防ぐ。ニチシノンはチロシン血症Ⅰ型の診断が確定したあと可及的速やかに開始するべきである。ニチシノンはチロシンの血中濃度を上昇させるので、角膜におけるチロシン結晶の形成を予防するため、フェニルアラニンやチロシン制限食を診断後速やかに開始するべきである。血中フェニルアラニン濃度が低くなりすぎた場合(<20μmol/L)、天然の蛋白質を食事に追加すべきである。ニチシノン登場以前のチロシン血症Ⅰ型に対する唯一の根治療法は肝移植であったが、現在では発症時に重度肝不全を認める小児例、ニチシノンに不応である小児例、もしくは肝組織に悪性化の所見を認める小児例にのみ行われるべきとされている。
一次症状の予防:
ニチシノンによる治療は診断確定後すみやかに行われるべきである。
二次合併症の予防:
腎尿細管ファンコーニ症候群に続発するカルニチン欠乏、骨粗鬆症、くる病を早期から治療する。
定期検査:
チロシン血症Ⅰ型患者において、ルーチンの定期検査に関するガイドラインが発行されている。
避けるべき薬物/環境:
不適切な蛋白摂取は避けるべきである。
リスクのある親族の検査:
チロシン血症Ⅰ型患児に続く同胞は全員、早期診断および治療を行えるように、尿中・血中サクシニルアセトンを出生後可及的速やかに測定するべきである。家系内の病原性変異が判明している場合、リスク妊娠に対して出生前の分子遺伝学的検査が考慮されることがある。
妊娠管理:
妊娠中にニチシノンを使用したデータはほとんどない。しかし、治療量のニチシノンを使用した少なくとも2人の女性から出生した児は健康であった。
遺伝カウンセリング
チロシン血症Ⅰ型は常染色体劣性遺伝性疾患である。受胎時に罹患者の同胞が罹患している確率は25%、無症候性キャリアである確率は50%、罹患もしておらずキャリアでもない確率は25%である。リスクのある家族に対する保因者診断やリスク妊娠における出生前診断は、家系での病原性変異が同定されている場合可能である。
診断
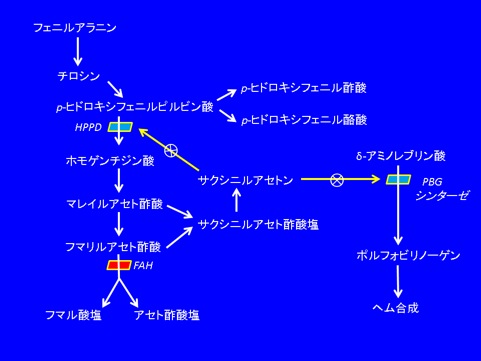
チロシン血症Ⅰ型はフマリルアセト酢酸ヒドラーゼ(fumarylacetoacetate hydrolase, FAH)(EC 3.7.1.2)の欠損による。(図1および「病態生理」を参照)
図1 チロシン分解経路
示唆的な所見
以下のような新生児スクリーニング結果、臨床徴候、支持的な検査所見を認めた場合にチロシン血症Ⅰ型を疑うべきである。
新生児スクリーニング
- 新生児ろ紙血からタンデム質量分析計により直接サクシニルアセトンを測定する。サクシニルアセトンの存在は、チロシン血症Ⅰ型に特徴的である。
- 血中チロシンまたはメチオニン濃度の上昇は肝疾患を示唆するが、その原因はさまざまである。チロシン血症Ⅰ型と診断するには、さらに血漿または尿中サクシニルアセトンの定量化を行うべきである。
- 最初の新生児スクリーニング検査で血中チロシンおよびメチオニン濃度の上昇はほんのわずかか、もしくは正常なことがある。
- 新生児スクリーニング検査でみられるチロシン濃度の上昇は、新生児期の一過性高チロシン血症、チロシン血症Ⅱ型/Ⅲ型、もしくはその他の肝疾患でも認めうる。
- メチオニン濃度の上昇は肝機能障害、メチオニン代謝異常、もしくはホモシスチン尿症を示唆しうる(シスタチオニンβ合成酵素欠損を原因とするホモシスチン尿症を参照)。
- δ-ALA脱水酵素(PBGシンターゼ)酵素活性はカナダ・ケベック州の新生児スクリーニングプログラムで測定されている。それから、見かけ上δ-ALA脱水酵素欠損症である乳児の尿でサクシニルアセトンを測定する。
臨床徴候(未治療患者)
- 乳児期早期の重症肝疾患
- 生後6ヶ月を超える小児では、腎疾患、くる病、神経性クリーゼの兆候
治療を受けていない小児患者では、1~7日間持続する神経性クリーゼを反復することがある。クリーゼでは、精神状態の変化、腹痛、末梢性ニューロパチー、および/もしくは人工換気を必要とする呼吸不全などを認める。
支持的な検査所見
血中サクシニルアセトン濃度の上昇および尿中排泄の増加
注:(1)肝不全/重症腎疾患患児におけるサクシニルアセトンの尿中排泄の増加はチロシン血症Ⅰ型の特徴である。(2)多くの検査機関は、尿中有機酸分析で特にサクシニルアセトンの測定を依頼するように要求している。
- 血漿チロシン・メチオニン・フェニルアラニン濃度の上昇
注:(1)乳児患者の血漿チロシン濃度は、臍帯血中および新生児期は正常なこともある。(2)血漿チロシン濃度上昇は、肝の障害もしくは未成熟の非特異的な指標にもなりうる。たとえば、希釈していないヤギ乳など高蛋白乳を摂取している乳児において認められる。
- 尿中チロシン代謝物の上昇
尿中有機酸分析でp-ヒドロキシフェニルピルビン酸、p-ヒドロキシフェニル乳酸、p-ヒドロキシフェニル酢酸が検出される。 - 肝臓や循環赤血球内のサクシニルアセトンによってδ-アミノレブリン酸(δ-ALA)脱水酵素が阻害され、δ-ALA化合物の尿中排泄が増加する。
- 肝機能の変化(未治療患者)
- 血清α-フェトプロテインの著明な上昇(平均160,000ng/mL)(正常:1~3ヶ月の乳児では1000ng/mL未満、3ヶ月~18歳では12ng/mL未満)
- プロトロンビン時間および部分トロンボプラスチン時間の延長
注:(1)チロシン血症Ⅰ型における血清α-フェトプロテイン(AFP)およびプロトロンビン時間/部分トロンボプラスチン時間(PT/PTT)の変化は非特異的な肝疾患よりも重度で、しばしば初発所見となる。(2)トランスアミナーゼおよびビリルビンの上昇はあったとしてもほんのわずかである。(3)血清AFP濃度正常およびPT/PTT正常の場合、肝疾患の原因がチロシン血症Ⅰ型である確率は低い。
診断の確定
チロシン血症Ⅰ型の診断は、特徴的な生化学的所見(血中および尿中サクシニルアセトン濃度の上昇、血漿チロシン・メチオニン・フェニルアラニン濃度の上昇、尿中のチロシン代謝物およびδアミノレブリン酸化合物濃度の上昇)および/または分子遺伝学的検査でFAH遺伝子両アレル病原性変異を同定することによって確定する(表1を参照)。
分子遺伝学的検査
単一遺伝子検査 まずはFAH遺伝子のシークエンス解析を行う。病原性変異が1つしか同定されない、もしくは1つも同定されない場合、続いて標的遺伝子の欠失/重複解析を施行する。
- アシュケナージ系ユダヤ人では、まずp.Pro261Leuのターゲット解析を行うことができる。これはアシュケナージ系ユダヤ人において病原性変異の99%以上を占める。
- フランス系カナダ人において、c.1062+5G>A(IVS12+5 G>A)は病原性変異の87.9%を占める。
- 米国において、主な4個のFAH遺伝子病原性変異、c.1062+5G>A(IVS12+5 G>A)、c.554-1G>T(IVS6-1 G>T)、c.607-6T>G(IVS7-6 T>G)、p.Pro261Leuはチロシン血症Ⅰ型で認める病原性変異の約60%を占める。
表1 チロシン血症Ⅰ型で用いられる分子遺伝学的検査
| 遺伝子1 | 検査方法 | 検査方法によって同定された変異2を有する発端者の比率 |
|---|---|---|
| FAH | シークエンス解析3 | >95% |
| 標的遺伝子の欠失/重複解析4 | 不明 巨大欠失が報告されている5 |
- 染色体座位と蛋白については、表A「遺伝子・データベース」を参照。
- この遺伝子で同定されるアレル変異に関する情報については、「分子遺伝学」の項を参照。
- シークエンス解析では、良性の変異、良性と考えられる変異、臨床的意義が不明の変異、病原性と考えられる変異、病原性変異が検出される。病原性変異には、小さな遺伝子内欠失・挿入、ミスセンス変異、ナンセンス変異、スプライス部位変異が含まれるが、典型的にはエクソンや遺伝子全体の欠失/重複は検出できない。シークエンス解析の結果の解釈について考慮すべき問題はこちらをクリック。
- 標的遺伝子の欠失/重複解析は、遺伝子内の欠失/重複を同定する。用いられる可能性のあるさまざまな方法には、この遺伝子/染色体領域を含んだ定量PCR、ロングレンジPCR、MLPA(multiplex ligation-dependent probe amplification)法、単一エクソンの欠失/重複を同定する標的遺伝子マイクロアレイ(gene-targeted microarray)などがある。
- Parkら(2009)はFAH遺伝子を含む巨大欠失を報告した。
臨床的特徴
臨床像
新生児スクリーニングで見つからなかったチロシン血症Ⅰ型患児は通常乳児期早期に重症肝疾患、もしくは乳児期後期に肝機能障害・著しい腎障害・成長障害・くる病で発症する。成長障害は慢性疾患に伴う経口摂取不良、肝機能障害、慢性腎疾患による。未発見および未治療の場合は通常10歳以前に死亡するが、典型的には肝不全、神経性クリーゼ、肝細胞癌による。
- 肝病変
生後6ヶ月以前に発症した未発見/未治療の患児では、典型的には凝固因子の合成障害からはじまる急性肝不全がみられる。PTおよびPTTが著明に延長し、ビタミンKの投与によっても補正されない。第Ⅱ、Ⅶ、Ⅸ、?、?因子濃度は低下する。第ⅤおよびⅧ因子の濃度は保たれる。逆説的ではあるが、血清トランスアミナーゼの上昇はほんの僅かなことがある。ほとんどの重症肝疾患ではPTやPTTの延長とともに著明なトランスアミナーゼや血清ビリルビンの上昇を認めるのに対し、血清ビリルビン濃度は正常か、上昇はあってもわずかである。認められる肝機能の乖離は、障害された肝細胞が細胞死しないためと説明されることがある。
この早期相から、腹水・黄疸・消化管出血を伴う肝不全へと進行しうる。患児は"蒸したキャベツ"もしくは"腐ったマッシュルーム"と形容される特徴的な臭いを認めることがある。ときに持続性の低血糖を呈する。一部は高インスリン血症を認める。その他、慢性的な軽度のアシドーシスを認める。未治療の乳児は初期症状から数週/数ヶ月以内に肝不全で死亡する可能性がある。 - 腎尿細管病変
より慢性の経過をとる未治療例では、症状は生後6ヶ月以降に出現する。腎尿細管病変は主要な症候である。腎尿細管機能障害は、汎アミノ酸尿、リン酸塩の喪失、そして多くに尿細管性アシドーシスを認めるファンコーニ様症候群(Fanconi-like renal syndrome)などがある。くる病はリン酸塩の持続的な腎からの喪失によると考えられている。血清カルシウム濃度は通常正常である。 - 神経性クリーゼ
未治療の患児では、年長の急性間欠性ポルフィリン症患者でみられるのと似た神経性クリーゼを反復することがある。これらのクリーゼでは、精神状態の変化、腹痛、末梢性ニューロパチー、人工換気を必要とする呼吸不全などが認められる。クリーゼは1~7日間持続しうる。反復する神経性クリーゼはしばしば気付かれていない。
- Mitchellら(1990)は、フランス系カナダ人の未治療小児患者の42%はそのようなエピソードを経験していると報告した。
- van Spronsenら(1994)による国際的な調査により、未治療患児の死亡の10%は神経性クリーゼの期間に起こっていることが報告された。
- 肝細胞癌
ニチシノンや低チロシン食による治療を受けていない患児や、急性発症の肝不全を克服した患児は、肝細胞癌の発症およびそれによる死亡リスクが高い。 - 未治療患児の生存率
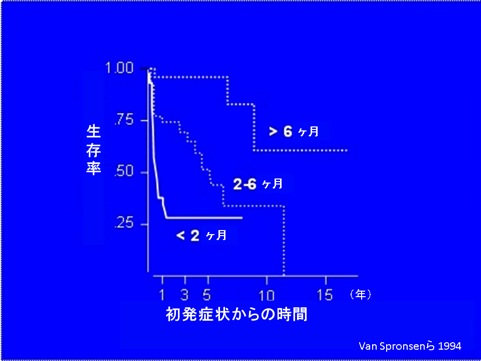
生後2ヶ月以前に診断された未治療患児の2年生存率は29%である。
- 診断が生後2~6ヶ月の場合、2年生存率は74%、生後6ヶ月以降の場合は96%である。
- 5年以上経過した後では、生後2ヶ月~6ヶ月の間に診断された群の生存率は約30%で、生後6ヶ月以降に診断された群の生存率は約60%に低下する(図2)。
図2 1992年以前の小児チロシン血症患者の生存率
治療を受けたチロシン血症Ⅰ型患者
ニチシノンの投与を受けた小児チロシン血症Ⅰ型患者の経過は未治療患児とは異なる。ニチシノンおよび低チロシン食による治療を受けた2歳未満の患児は、低チロシン食単独の患児に比べて著明な改善がみられる。ニチシノンおよび低チロシン食により、90%を超える生存率、正常な成長、肝機能の改善、肝硬変の予防、尿細管性アシドーシスの是正、二次性くる病の改善を認める。
- 治療を受けた患児でみられる神経性クリーゼは、ニチシノンの長期にわたる中止期間と常に相関する。
- 急性肝不全では、ニチシノンを開始する前および開始した当初は支持療法を必要とする。一般的にニチシノンを開始して1週間以内に改善が認められる。
- 角膜結晶(Corneal crystals)
ニチシノンはチロシン代謝経路を阻害し、サクシニルアセトンは生成されないため組織チロシン濃度は上昇する。600mol/Lを超える血中チロシン濃度はチロシン沈着のリスクとなり、両側・線状・分枝状の上皮下角膜混濁によって羞明、掻痒感、視覚過敏(sensitive eyes)をきたす。チロシン濃度が低下すると結晶は消失する。 - 肝細胞癌
HolmeやLindstedt(2000)ら、van Spronsenら(2005)によってニチシノン開始数年後に肝細胞癌を発症した症例が報告されているが、2歳以前にニチシノンの投与を受けた患児で10歳までに肝細胞癌を発症するのは5%未満と推定されている。
新生児スクリーニング検査にチロシン血症Ⅰ型を含めているケベック州では、チロシン血症Ⅰ型の症状で入院した患者はおらず、日齢30以前からニチシノンの投与を受けた患者では肝細胞癌の報告はない。この群で最も長い治療期間は12年である。
病態生理
フマリルアセト酢酸ヒドラーゼ(FAH)はチロシン代謝経路の最終酵素である(図1)。FAH(EC 3.7.1.2)の欠損はチロシン血症Ⅰ型を起こす。FAHが欠損すると、直接前駆体であるフマリルアセト酢酸(FAA)は、
- 肝細胞に蓄積し、細胞の障害やアポトーシスを起こすようである(Endo & Sunによる動物実験で認められた)。
・サクシニルアセト酢酸やサクシニルアセトンに変化する。サクシニルアセトンは以下の肝酵素の活性を阻害する。
- パラヒドロキシフェニルピルビン酸ジオキシゲナーゼ(p-HPPD)を阻害し、血漿チロシン濃度が上昇する。
- PBGシンターゼを阻害し、(1)肝臓や循環赤血球中のδ-ALA脱水酵素活性が低下する。(2)ヘムの合成が減少する。(3)急性の神経学的エピソードを起こすことがあるδ-ALAが増加する。(4)尿中へのδ-ALA排泄が増加する。
遺伝子型と臨床型の関連
一般的に、臨床症状と遺伝子型の間に相関は認められない。同じ遺伝子型の非血縁者と同様に、同一家系内でも急性型、慢性型ともにみられる。
この臨床的な多様性を説明できる1つの機序に遺伝子変異の復帰(gene reversion)がある。慢性型のチロシン血症Ⅰ型患者の肝臓より摘出された肝結節において、免疫学的にFAH蛋白陽性の細胞やFAHの酵素活性が認められている。これらの一見"正常な"細胞は遺伝子変異の復帰(gene reversion)、すなわち体細胞分裂時に生殖細胞の病原性変異が正常な遺伝子配列に自然に修復される(すなわちreversionもしくは"back-mutation[復帰突然変異]")ことによって生じる。
病原性変異の効果を抑制し、遺伝子発現は正常もしくは正常に近い自然発生的な体細胞変異も報告されている。これは変異した配列の真の復帰であり、母体由来細胞のコロニー形成もしくは融合によるものではない。フマリルアセト酢酸(FAA)の蓄積によるアポトーシスのリスクがもはやないため、 "正常な"(すなわち復帰した)細胞は選択的成長優位性(selective growth advantage)を示す。生化学所見や臨床所見が軽微な未治療の慢性チロシン血症Ⅰ型患者において、肝結節の多くは遺伝子の変異復帰による"正常な"細胞コロニーから構成されている。しかし、復帰のない変異細胞(non-revertant mutant cells)はフマリルアセト酢酸を持続的に生成し、肝細胞癌のリスクを高める。
生後4ヶ月のベルギー人の重症肝疾患患者で、稀で非典型的なチロシン血症Ⅰ型の病型が報告されている。α-フェトプロテインは著明に上昇し、PT・PTTは延長していたが、尿中サクシニルアセトンは検出されなかった。フマリルアセト酢酸(FAH)蛋白や活性は減少・低下しているが、欠損はしていなかった。c.103G>A(p.Ala35Thr)という独特な病原性変異のホモ接合体が同定された。
同じように、血中/尿中サクシニルアセトンは検出されないが慢性肝疾患および肝細胞癌を発症した3人の同胞例において、FAH酵素活性欠損が認められた。家族は中東出身で、それぞれの患児はFAH遺伝子にc.424A>Gのホモ接合体を有していた。
命名
以前用いられていた病名にチロシノーシス(tyrosinosis)などがある。
発生率
新生児スクリーニングが行われていない地域では、チロシン血症Ⅰ型は約100,000-120,000出生に1人に認められている。臨床症状に一貫性はなく混同されやすいため、生存している患者の50%未満しか診断されていないと推測されている。
米国では、キャリア頻度は1:150~1:100と推定されている。
創始者効果によって病原性変異が高頻度であるため、チロシン血症Ⅰ型の頻度が予想より高い地域が世界に2つある。
- スカンジナビア諸国[c.1062+5G>A(IVS12+5 G>A), p.Gly337Ser, p.249HisfsTer5.5]やフィンランド[p.Trp262Ter]では、発生率はそれぞれ74,000出生に1人、60,000出生に1人と推定されている。
- カナダのケベック州では、フランス人開拓者の入植による創始者効果が存在する。c.1062+5G>A(IVS12+5 G>A)変異はこの集団におけるアレル変異の87%を占める。ケベック州における発生率は16,000出生に1人である。ケベック州のサグネ・ラック・サン・ジャン地域では、1,846出生に1人である。新生児スクリーニング検査に基づくケベック州全体のキャリア頻度は66人に1人である。サグネ・ラック・サン・ジャン地域におけるキャリア頻度は16~20人に1人である。
遺伝学的関連(アレル)疾患
このGeneReviewで論じられている以外に、FAH遺伝子変異と関連する臨床型は知られていない。
鑑別診断
小児で以下の所見のいずれかを認める場合、チロシン血症Ⅰ型を評価すべきである(表2)。
表2 初発所見によるチロシン血症Ⅰ型乳児の鑑別疾患
| 初発所見 | 鑑別疾患 |
|---|---|
| 高チロシン血症 | ・未成熟な肝臓 ・高蛋白食1, 2 ・チロシン血症Ⅱ型(OMIM276600) ・チロシン血症Ⅲ型(OMIM276710) ・その他の肝疾患 |
| 高メチオニン血症 | ・ホモシスチン尿症 ・メチオニン代謝異常 ・その他の肝疾患 |
| 肝疾患 | ・ガラクトース血症 ・遺伝性果糖不耐症 ・フルクトース-1,6-ビスホスファターゼ欠損症(OMIM229700) ・ニーマン・ピック病C型 ・ウィルソン病 ・新生児ヘモクロマトーシス(OMIM231100) ・血球貪食性リンパ組織球症 ・ミトコンドリア病 ・先天性グリコシル化異常症 ・トランスアルドラーゼ欠損症(OMIM606003) ・アセトアミノフェン中毒 ・細菌感染症(敗血症、サルモネラ、結核) ・ウイルス感染症(CMV、A型/B型肝炎、ヘルペスなど) ・キノコ中毒3 ・薬草3 ・特異な薬物反応、毒素、血管性/虚血性、炎症性3 |
| 腎疾患 | ・ロウ症候群 ・シスチン症 ・尿細管性アシドーシス ・ファンコーニ症候群 |
| くる病 | ・低ホスファターゼ症 ・ビタミンD欠乏(栄養性/遺伝性) ・低リン血症性くる病 ・ビタミンD依存性くる病 ・ファンコーニ症候群 |
| 神経性クリーゼ | ・脳出血/浮腫 ・細菌性/ウイルス性髄膜炎 ・高張性脱水 ・急性間欠性ポルフィリン症 |
- Techakittirojら(2005)
- 希釈していないヤギ乳
- Bansal & Dhawan(2004)
チロシン血症Ⅱ型はチロシンアミノトランスフェラーゼ(TAT)(EC2.6.1.5)の欠損によって起こる。チロシン血症Ⅱ型の診断は以下による。
- 血漿チロシン濃度は典型的には500μmol/Lを超え、1000μmol/Lを超えることもある(その他のアミノ酸の濃度は正常)。
- 尿中有機酸分析で、p-ヒドロキシフェニルピルビン酸、p-ヒドロキシフェニル乳酸、p-ヒドロキシフェニル酢酸の排泄が増加し、N-アセチルチロシンや4-チラミンが少量検出される。
手掌や足底に痛みはあるが掻痒感はない角化性局面を認める。足趾底面に過角化を伴った著明な黄色の肥厚を認めることがある。眼病変には難治性の偽樹枝状角膜炎がある。発達遅滞はよく認められるが、発達遅滞や神経症状の報告が診断バイアスによるのかは不明である。
所見はチロシンやフェニルアラニンの制限食で改善する。
チロシン血症Ⅲ型はチロシン血症のなかでもっとも稀な疾患であり、p-ヒドロキシフェニルピルビン酸ジオキシゲナーゼ(EC.1.13.11.7)の欠損によって起こる。血漿チロシン濃度は350~650μmol/Lを示す。4-ヒドロキシフェニルピルビン酸、4-ヒドロキシフェニル乳酸、4-ヒドロキシフェニル酢酸の排泄が増加している。正確な排泄量は摂取した蛋白量によって変わる。
見つかっている患者は少なく、臨床型は明らかではない。最初の患者は知的障害もしくは運動失調のため治療対象となる。その他の者はルーチンのスクリーニング検査で見つかっている。チロシン血症Ⅱ型患者のように、Ⅲ型患者は肝病変を有さないが皮膚/眼病変は認められる。チロシン血症Ⅲ型が本当に認知の遅れと相関するのかどうか、その相関が診断バイアスによるのかどうかは依然として不明である。
低フェニルアラニン・チロシン食により血漿チロシン濃度を低下させることができる。
臨床的マネジメント
初期診断後の評価
新生児スクリーニングに基づいてチロシン血症Ⅰ型と診断された患者の疾患の広がりやニーズを把握するために、以下の評価が推奨される(表3を参照)。
- 血小板数を含めた血算
- 血清電解質
- 肝機能(PT, PTT, AST, ALT, GGT, 血清ビリルビン, ALP, 血清AFP)
臨床症状に基づいて(表4[pdf]を参照)診断された小児では、上記に加えて以下の評価を行うべきである。
- 肝腺腫/結節の評価のため(造影)CT/MRIによるベースラインの腹部画像検査
- くる病の有無の評価のため手関節X線
すべての患者は臨床遺伝専門医に紹介する。
病変に対する治療
治療ガイドラインが発行されている。米国版はChinsky JMらの文献を、欧州版はde Laetらの文献などに基づいている。
肝不全急性期の治療 換気補助、適切な輸液管理、出血傾向に対する血液製剤による補正を必要とすることがある。
ニチシノン(オーファディン®) 2-(2-ニトロ-4-トリフルオロ-メチルベンゾイル)1,3-シクロヘキサンジオン(NTBC)はチロシン分解経路の第2段階であるパラヒドロキシフェニルピルビン酸ジオキシゲナーゼ(parahydroxyphenylpyruvic acid dioxygenase, p-HPPD)を阻害し、フマリルアセト酢酸の蓄積とそのサクシニルアセトンへの変換を防ぐ(図1)。
- ニチシノンはチロシン血症Ⅰ型の診断が確定したあと可及的速やかに開始するべきである。
- ニチシノンの一般的な処方量は1.0mg/kg/日である。個人によって必要量は異なる。用量は、理論的にp-HPPD活性を99%以上阻害する血中ニチシノン濃度40~60μmol/Lの間に保つよう調節すべきである。まれに、サクシニルアセトン排泄抑制のためより高い血中ニチシノン濃度(70μmol/L)が必要となることがある。血中ニチシノン濃度が治療域内である限り、尿中サクシニルアセトン濃度を測定する必要はない。
- ニチシノンは典型的には1日2回に分服する。しかし、半減期が長いため(50-60時間)、1歳以上で安定している患者は1日1回で維持することもある。
- ニチシノンのまれな副作用には、治療を必要としない一過性の血小板減少や好中球減少、より厳格な食事制限で血中チロシン濃度を低下させると軽快する羞明などがある。
低チロシン食 ニチシノンはチロシンの血中濃度を上昇させるため、角膜にチロシン結晶が形成されないよう低チロシン食とする必要がある。
- 食事管理は診断後すぐに開始するべきで、低蛋白の菜食料理やTyrex®(Ross)/Tyros-1(Mead Johnson)のような医療用ミルクを用いて、フェニルアラニンやチロシンを制限した栄養学的に完全な食事を摂取するべきである。
- フェニルアラニンやチロシンの必要量は相互依存的で、患者ごとや同じ患者でも成長率・エネルギーや蛋白の摂取量・健康状態によって異なる。適切な食事管理により、血漿チロシン濃度は年齢にかかわらず300-500μmol/Lとするべきである。血漿フェニルアラニン濃度は20-80μmol/L(0.3-1.3mg/dL)とするべきである。血中フェニルアラニン濃度が低くなりすぎた場合(20μmol/L未満)、蛋白質をミルクや食品に追加すべきである。
肝移植 チロシン血症Ⅰ型の治療でニチシノンが登場する前は、肝移植が唯一確立された治療法だった。
- 肝移植の適応は以下のような場合に制限するべきであるとしている。(1)発症時に重篤な肝不全を呈し、ニチシノンに抵抗性を示す場合。(2)肝組織で悪性所見を認める場合。
- 肝移植は長期の免疫抑制剤を必要とする。年少児における肝移植に関連する死亡率は10%以上である。
- 血漿・尿中のサクシニルアセトンによる持続的な尿細管・糸球体機能障害を予防するため、レシピエントに対する低用量(0.1mg/kg/日)ニチシノンも有用である。
一次症状の予防
ニチシノン(オーファディン®)による治療は診断が確定したら可及的速やかに開始するべきである。
二次合併症の予防
腎尿細管ファンコーニ症候群に続発するカルニチン欠乏は骨格筋の筋力低下を起こすため、カルニチン欠乏を認めた場合に治療できるように血清カルニチン濃度を測定すべきである。
尿細管障害による骨粗鬆症・くる病に対する治療は、アシドーシスの補正、カルシウム・リンのバランスの維持、25-ヒドロキシ-ビタミンDの投与を行う。
定期検査
チロシン血症Ⅰ型患者の典型的な管理では、以下の指標を頻回に評価する(表3)。
表3 ガイドラインで提唱されている新生児スクリーニングで診断されたチロシン血症Ⅰ型患者のモニタリング
| 評価 | 治療開始時(ベースライン ) | はじめの6ヶ月 | 治療開始6ヶ月以降:6-12ヶ月ごと | 治療開始2年以降:6-12ヶ月ごと | 臨床的に適応があるとき | ||
|---|---|---|---|---|---|---|---|
| 1ヶ月ごと | 3ヶ月ごと | ||||||
| チロシン血症Ⅰ型のマーカー | 血漿メチオニン・フェニルアラニン・チロシン濃度 | X | X | X | X | またはX | |
| 血中/尿中サクシニルアセトン | X | X(尿) | X | またはX | |||
| 血中ニチシノン濃度 | X | X | X | またはX | |||
| 血算 | ヘモグロビン、ヘマトクリット、白血球数、血小板数 | X | X | X | X | またはX | |
| 肝臓の評価 | 血清AFP濃度 | X | X | X | X | X | |
| PT/PTT | X | X(正常になるまで) | |||||
| ビリルビン | X | ||||||
| ALT/AST/GGT | X | X(正常になるまで) | X | ||||
| アルカリホスファターゼ | X | X(正常になるまで) | X | X | |||
| CTもしくはMRI1 | X | ||||||
| 腎検査 | BUN, クレアチニン | X | |||||
| 尿:PO4, Ca, 蛋白/クレアチニン比 | X | ||||||
| 骨格系の評価 | 手関節X線(くる病) | X | |||||
AFP=α-フェトプロテイン
ALT/AST=アラニンアミノ基転移酵素/アスパラギン酸アミノ基転移酵素
BUN=血中尿素窒素
GGT=γ-グルタミルトランスフェラーゼ
PT/PTT=プロトロンビン時間/部分トロンボプラスチン時間
- 造影MRIで肝腺腫/結節や腎臓のサイズを評価する。
臨床所見に基づき診断された小児のモニタリングについては表4を参照(pdf)。
他に推奨されている管理方法については、de Laetらの報告(2013)を参照。

避けるべき薬物/環境
不適切な蛋白摂取を避ける。
リスクのある親族の検査
患児の同胞は一見無症状であっても、治療や予防を迅速に開始できるよう、なるべくすみやかに評価を行うことがのぞましい。
評価方法には、
- 家系内のFAH遺伝子変異が既知である場合、分子遺伝学的検査を行う。出生前にリスクのある同胞の遺伝学的状況を明らかにするために、出生前診断を行うことができる。
- 出生前診断が行われていない場合、可及的すみやかに血中/尿中サクシニルアセトン分析を行う。これにより、出生後に遺伝学的検査の結果をタイミングよく得られない場合に、すみやかな治療を開始することができる。
- 家系内のFAH遺伝子変異が既知である場合、年上の健康な同胞の尿中サクシニルアセトン分析を考慮する。
遺伝カウンセリングとして扱われるリスクのある親族への検査に関する問題は「遺伝カウンセリング」の項を参照のこと。
妊娠管理
ヒトで妊娠中にニチシノンを使用したデータはほとんどない。妊婦に有害事象は起こらないだろうと推測されているが、チロシン代謝が変化するため胎児の発達に対するリスクになる可能性がある。
治療量のニチシノンを使用した少なくとも2人の女性から出生した児は健康であった。
- 1例では、罹患女性から産まれた児は健康で2歳半まで正常に発達している。
- もう1例では、罹患女性から産まれた児は罹患者であった。児の生後7ヶ月までの成長発達は正常である。報告者らは、妊娠中に母親がニチシノン投与を受けることで、患児は胎内で肝障害から守られるのだろうと推察している。
妊娠中における薬剤投与に関するさらなる情報についてはwww.mothertobaby.orgを参照のこと。
研究中の治療法
さまざまな疾患に関する臨床試験に関する情報はClinicalTrials.govを参照のこと。注:この疾患に対する臨床試験は行われていない可能性がある。
その他
ニチシノンが登場する前は、移植以外の有用な治療法はフェニルアラニン・チロシン制限食のみであった。効果はわずかなもので、神経性クリーゼの反復や肝疾患の進行が起こる。平均生存期間は10年未満である。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
チロシン血症Ⅰ型は常染色体劣性遺伝性疾患である。
患者家族のリスク
発端者の両親
- 患児の両親は必然的にヘテロ接合体保有者(すなわち1つのPAH病原性アレル変異のキャリア)である。
- ヘテロ接合体保有者(キャリア)は無症状であり、発症リスクはない。
発端者の同胞
- 受胎時に罹患者の同胞が罹患している確率は25%、無症候性キャリアである確率は50%、罹患もしておらずキャリアでもない確率は25%である。
- ヘテロ接合体保有者(キャリア)は無症状であり、発症リスクはない。
発端者の子
- チロシン血症Ⅰ型患者の子どもは必然的にFAH変異のヘテロ接合体保有者(キャリア)である。
発端者の他の家族
- 両親の同胞がFAH遺伝子変異のキャリアである確率はそれぞれ50%である。
保因者診断
リスクのある親族に対する保因者診断を行う前に、家系内の病原性変異を同定する必要がある。
遺伝カウンセリングに関連した問題
早期診断・治療を目的としたリスクのある親族の検査についての情報は、「臨床的マネジメント」「リスクのある親族の検査」を参照のこと。
家族計画
- 遺伝学的リスク評価、保因者診断、および出生前診断の利用について話し合いを行う最適な時期は妊娠前である。
- 罹患者、キャリア、もしくはキャリアのリスクがある若年成人に対して遺伝カウンセリング(潜在的な子どもへのリスクや出産方法の選択肢に関する話し合いなど)を申し出ることがのぞましい。
DNAバンクは(主に白血球から調整した)DNAを将来利用することを想定して保存しておくものである。検査技術や遺伝子、アレル変異、あるいは疾患に対するわれわれの理解が将来さらに進歩すると考えられるので、患者のDNA保存を考慮すべきである。
出生前診断および着床前診断
分子遺伝学的検査
家系内で罹患者のFAH病原性変異が明らかとなっている場合、リスク妊娠に対して出生前診断や着床前診断を行うことができる。
生化学検査
25%のリスクがある妊娠で、出生前診断は、通常はおよそ妊娠15-18週に行う羊水穿刺によって得られた羊水中にサクシニルアセトンを検出することで行うことができる。羊水中にサクシニルアセトンを検出すれば診断できるが、偽陰性も報告されている。そのため、この方法は安定同位体検出によって常に低濃度のサクシニルアセトンを検出することができる検査機関でのみ用いられるべきである。生化学検査にはこれらの問題があるため、出生前診断の方法として分子遺伝学的検査のほうがのぞましい。
更新履歴
- Gene Reviews著者: Lisa Sniderman King, MSc, CGC, Crisine Trahms, MS, RD, and C Ronald Scott, MD.
日本語訳者: 和田宏来 (県西総合病院小児科/筑波大学大学院小児科)
Gene Reviews 最終更新日: 2017.5.25.日本語訳最終更新日: 2017.7.24(minor revision: 2017.10.30)(in present)
![]()