色素性乾皮症
(Xeroderma Pigmentosum)
Kenneth H Kraemer, MD and John J DiGiovanna, MD.
日本語訳者: 窪田美穂(ボランティア翻訳者),櫻井晃洋札幌医科大学附属病院遺伝子診療室)
Gene Review 最終更新日:2016.9.29,日本語訳最終更新日:2017.1.15
要約
疾患の特徴
色素性乾皮症(xeroderma pigmentosum, XP)の特徴は以下である.
- 日光過敏症(患者の60 %未満には,わずかな日光曝露で,水疱や持続性紅斑を伴う重度の日焼けが起きる).大多数の患者では,2歳以前に,顔面に顕著なそばかす様の色素沈着が生じる.
- 日光誘発性の眼症状(羞明,角膜炎,眼瞼の皮膚萎縮)
- 日光誘発性の皮膚腫瘍(基底細胞癌,扁平上皮癌,黒色腫)のリスクは極めて高い.
患者の約25 %に神経症状(後天性小頭症,深部腱反射の減弱もしくは消失,進行性の感音難聴,進行性の認知障害)が現れる.最も多い死因は皮膚癌,神経変性,内臓腫瘍である.神経変性を伴うXP患者の死亡年齢(中央値)は29歳であり,神経変性を伴わない患者(37歳)よりも早いことがわかっている.
診断・検査
XPの診断は臨床所見や家族歴に加え,DDB2遺伝子,ERCC1遺伝子,ERCC2遺伝子,ERCC3遺伝子,ERCC4遺伝子,ERCC5遺伝子,POLH遺伝子,XPA遺伝子,XPC遺伝子の両アレルに病原性多型が同定されることに基づく.
臨床的マネジメント
症状の治療:日光性角化症などの前癌状態の小さな皮膚病変には液体窒素による凍結療法を行う.これよりも大きな病変には5-フルオロウラシル軟膏やイミキモド・クリーム(imiquimod)などを用いた局所療法を行う.稀に,採皮刀による薄片切除術や皮膚剥離術が行われる.皮膚腫瘍は,(XP患者でない場合と同様に)電気乾固・掻爬術や外科的切除により治療可能である.再発性の皮膚癌や再発リスクの高い部位の皮膚癌への最適な治療はモース顕微鏡手術である.イソトレチノイン(isotretinoin)やアシトレチン(acitretin)の経口投与により新たな皮膚新生物の発生を抑えることができるが,多くの副作用がある.眼瞼,結膜,角膜の新生物には外科的治療を行う.角膜移植により,重度の角膜炎に起因する視力障害が改善することがある.難聴には補聴器を使用してもよい.
一次病変の予防:皮膚や眼が日光や紫外線にあたらないようにする.患者の自宅,学校,職場環境の紫外線量を測光露出計で測定し,環境紫外線量が高い場所を特定し,こうした場所をなくすとよい.
二次合併症の予防:必要に応じて,ビタミンDのサプリメントを摂取する.
経過観察:3~12ヶ月に1回の医師による皮膚の診察.定期的に眼科検査,神経学的検査,聴力検査を実施.
回避すべき薬剤・環境:日光や紫外線の人工光源への曝露,タバコの煙は避けるべきである
リスクのある近親者の検査:病原性多型がわかっているならば,リスクのある同胞に分子遺伝学的検査を行うことで早期診断が可能となり,幼児期から確実に日光防護を行うことができる.
妊娠管理:皮膚癌の予防として,レチノイド(イソトレチノイン,アシトレチン)の全身投与が行われる場合がある.こうした薬剤には催奇形性があることがわかっており,先天異常のリスクが高くなる.妊娠可能年齢にある女性がレチノイドを使用する場合,確実に避妊を行い,定期的に妊娠検査を受けること.
遺伝カウンセリング
XPの遺伝形式は常染色体劣性である.受精時に同胞が発症する確率は25 %,無症候性の保因者である確率は50 %,発症せず,保因者でもない確率は25 %である.血縁者に病原性多型が同定されたら,リスクのある血縁者への保因者診断や,リスクの高い妊娠への出生前診断を検討できる.
診断
XPが疑われる所見
皮膚,眼,神経系,家族歴に以下の所見を認める場合,色素性乾皮症を疑うべきである.
皮膚
- 急性日光過敏症(わずかな日光曝露で生じる水疱や持続性紅斑を伴った重度の日焼け)
- 2歳以前での顔面の顕著なそばかす様色素沈着(黒子)
- 1歳以前での皮膚癌
眼
- 顕著な結膜充血を伴う羞明
- 重度の角膜炎(角膜混濁や血管新生が生じることもある)
- 眼瞼への色素沈着の進行,睫毛消失
- 眼瞼の皮膚萎縮による眼瞼外反,眼瞼内反.重症例では眼瞼の完全な消失.
神経系
- 深部腱反射の減弱もしくは消失筋電図や神経伝導速度検査で軸索性(もしくは混合性)ニューロパチーがみつかることがある.
- 進行性感音難聴.聴力検査で早期の高音障害型難聴がみつかることがある.
- 後天性小頭症.脳のCTやMRIで,皮質の菲薄化と頭蓋骨の肥厚化を伴う脳室拡大を認めることがある.
- 進行性認知障害
家族歴
- 常染色体劣性に一致する遺伝形式
注:XPの家族歴がなくても,診断が不可能となるわけではない.
確定診断
発端者のXPの診断は,臨床所見や家族歴(「XPが疑われる所見」の項を参照)に加え,表1に記載された遺伝子の1つの両アレルに病原性多型が同定されることに基づいて行われる.
分子遺伝学的検査の手法には,単一遺伝子に対する検査を連続的に行う方法(serial single-gene testing),複数遺伝子パネル,より網羅的な全ゲノム解析がある.
単一遺伝子の連続検査.検査する遺伝子は,臨床徴候や患者が属する民族での相対頻度に基づいて選択する(表1,表2図1,DiGiovanna & Kraemer [2012]を参照).
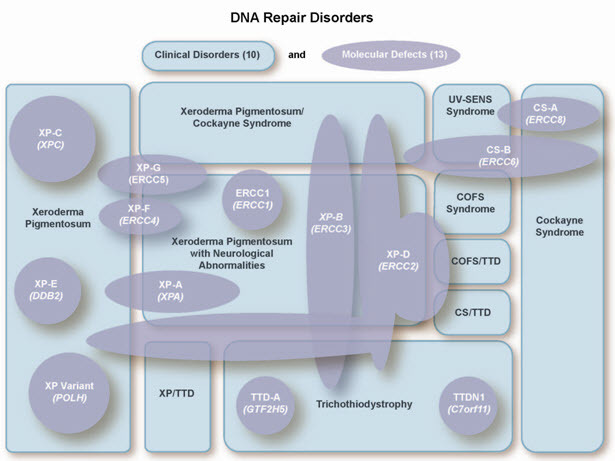
色素性乾皮症-コケイン症候群-裂毛症スペクトラムの遺伝型と表現系との関連.斜体の文字は変異が生じている遺伝子を指す.四角は表現型を指す.遺伝型と臨床型の相関は複雑であり,変異によってDNA修復や転写経路に異なる影響を及ぼすこともあるため,個々の患者の臨床像を相補性から予測することは難しい.
まず,その地域に存在する創始者バリアントの検査を考慮するとよい.
- XPA
- インド:c.335_338delTTATinsCATAAGAAA [Tamhankar et al 2015]
- 日本:c.390-1G>C(保因者頻度は1 %)[Hirai et al 2006]
- チュニジア:p.Arg228Ter [Messaoud et al 2010]
- XPC. 北アフリカ:c.1643_1644delTG [Soufir et al 2010,Hadj-Rabia et al 2013,Jerbi et al 2016]
- ERCC2 .イラク系ユダヤ人:p.Arg683Gln [Falik-Zaccai et al 2012]
- POLH
- チュニジア/北アフリカ:エクソン10欠失[Ben Rekaya et al 2014]
- 日本:c.490G>T(スプライス部位変異);p.Ser242Ter,p.Glu306Ter,c.1661delA[Masaki et al 2008]
- バスク/スペイン北部:c.764+1G>A[Calmels et al 2016]
複数遺伝子パネル(DDB2遺伝子,ERCC1遺伝子,ERCC2遺伝子,ERCC3遺伝子,ERCC4遺伝子,ERCC5遺伝子,POLH遺伝子,XPA遺伝子,XPC遺伝子やこのほかの関連がありそうな遺伝子)も検討すること(「鑑別診断」の項を参照).注:(1)複数遺伝子パネルに含まれる遺伝子や検査の精度は,検査機関によって異なっているだけでなく,時代とともに変化する.(2)複数遺伝子パネルに本稿で扱っている病態に関連のない遺伝子が含まれている場合もあるため,臨床医は最善のコストで遺伝的原因を確定できるように,どの検査を行うのが最適であるかを決定しなければならない.(3)こうした検査で用いられる手法には,配列解析,欠失・重複解析,配列解析を行わない検査がある.
より網羅的なゲノム解析.1つの遺伝子に対する検査や複数遺伝子パネル(DDB2遺伝子,ERCC1遺伝子,ERCC2遺伝子,ERCC3遺伝子,ERCC4遺伝子,ERCC5遺伝子,POLH遺伝子,XPA遺伝子,XPC遺伝子)でXPを示す所見をもつ患者の診断が確定しない場合,(実施可能であるならば),より包括的な全エクソーム解析(WES)や全ゲノム 解析(WGS)を考慮してもよい.こうした検査により,これまで検討していなかった診断が示されることもある(類似所見を生じさせる別の遺伝子変異など).ゲノム解析の結果の解釈の問題については,こちらを参照のこと.
表1.
色素性乾皮症の分子遺伝学的検査
| 遺伝子1 | 病原性多型ごとの患者の割合(%)2 | 検査法ごとの病原性多型3の検出割合 | |||
|---|---|---|---|---|---|
| 米国 | 日本 | 欧州 | 配列解析4 | 標的遺伝子の欠失・重複解析5 | |
| DDB2 | 3 % | 3 % | 15/15アレル6 | 不明7 | |
| ERCC1 | 稀 | 4/4アレル8 | 不明 | ||
| ERCC2 | 28 % | 5 % | 16 % | >99 % | 不明 |
| ERCC3 | 1 % | 0 % | 2 % | 8/8アレル9 | 不明 |
| ERCC4 10 | 0 % | 7 % | 3 % | ~99 % | 稀11 |
| ERCC5 | 3 % | 1 % | 9 % | 99 %超 | 不明 |
| POLH | 7 % | 25 % | 13 % | 25/40アレル;33/42アレル12 | 15/40アレル;9/42アレル12 |
| XPA | 9 % | 55 %13 | 20 % | 100 % | 不明 |
| XPC | 43 % | 3 % | 31 % | 7/12アレル14 | 5/12アレル14 |
- 染色体座位と蛋白については,表A「遺伝子・データベース」を参照.
- 頻度は,米国の106人のXP患者についてはBradford et al [2011]から,日本の291人のXP患者についてはMoriwaki & Kraemer [2001]から,英国(ロンドン)の診療所の89人についてはFassihi et al [2016]から得られた.
- 検出される病原性変異に関する情報については,「分子遺伝学」の項を参照されたい.
- 配列解析では,良性多型,良性と考えられる多型,臨床的意義が不明の多型,病原性と考えられる多型,病原性多型(変異)が検出される.病原性多型には,小さな遺伝子内の欠失・挿入,ミスセンス変異,ナンセンス変異,スプライス部位変異が含まれるが,エクソンや遺伝子全体の欠失・重複は検出できない.配列解析の結果の解釈についてはこちらを参照(検出されうる変異:既に報告されている病原性変異,病原性と推測されるが過去の報告がない変異,臨床的意義が不明なシークエンス変化,病的意義がないと考えられるが過去に報告がないシークエンス変化,既に報告されている病原性のないシークエンス変化.変異が検出されない場合に考えられる可能性:患者は解析した遺伝子に変異を有していない,患者は変異を有しているがシークエンス解析で検出できない).
- 標的遺伝子の欠失・重複解析では,遺伝子内の欠失や重複が検出できる.検査方法は,定量的PCR,ロングレンジPCR,MLPA(multiplex ligation-dependent probe amplification)法,単一エクソンの欠失や重複の検出を目的とする標的遺伝子マイクロアレイなどである.
- Nichols et al [1996], Rapi?-Otrin et al [2003], Fassihi et al [2016]
- 標的遺伝子の欠失・重複解析の検出率に関するデータは入手できなかった.
- Jaspers et al [2007], Kashiyama et al [2013]
- Oh et al [2006], Fassihi et al [2016]
- ERCC4遺伝子に病原性バリアントをもつ患者では,ファンコニ貧血の症状を伴う症例,XP-コケイン症候群複合型を呈する症例,もしくはXP-コケイン症候群-ファンコニ貧血の複合型を呈する症例が報告された[Bogliolo et al 2013,Kashiyama et al 2013].
- エクソン3欠失が報告された[Ahmad et al 2010].
- POLH遺伝子の多型のほとんどが配列解析で検出可能であるが,単一エクソン,もしくは複数エクソンの欠失も報告されている.欠失の頻度は,民族ごとに異なっている[Broughton et al 2002,Opletalova et al 2014].チュニジアのXP-V患者では,32アレルのうち32がエクソン10の欠失であった[Ben Rekaya et al 2014].
- 日本に多く,米国や欧州では稀.
- Chavanne et al [2000]
検査の特徴.検査の精度や特異性については,Clinical Utility Gene Card[Schubert et al 2014]を参照.
DNA修復異常のスクリーニングには,細胞での紫外線高感受性や不定期DNA合成(UDS),宿主細胞回復を調べる検査などの生細胞に対する機能検査が用いられるが,こうした検査は一般的に行われる検査ではない[Stefanini & Kraemer 2008,Kraemer & Ruenger 2012,Ruenger et al 2012].
研究のみを目的とした検査方法に関するこのほかの情報については,こちら(pdf)を参照.
臨床的特徴
臨床像
米国国立衛生研究所(NIH)でBradford et al [2011]が1971年から2009年まで106人のXP患者に対して行った長期的研究から得られた所見を以下に記す.これまでの研究からの引用も含む.
色素性乾皮症(XP)
- 皮膚.XP患者の約60 %には,わずかな日光曝露による急性の日焼け反応の既往がある.これ以外の40 %のXP患者では,過剰な日焼けは生じなかった[Sethi et al 2013].すべての患者の日光曝露部位の皮膚には,そばかす様の色素過剰斑が無数に現れる.皮膚症状の発症年齢(中央値)は1~2歳である[Kraemer et al 1987,Kraemer et al 1994].こうした異常が現れるのは日光曝露部位に限られる.
日光曝露が続くと皮膚は乾燥して羊皮紙様となり,色素沈着も亢進することから,色素性乾皮症(「乾燥した色素沈着した皮膚」)と名付けられた.大多数のXP患者が乾燥症(皮膚乾燥)と多形皮膚萎縮症(色素沈着過剰/色素欠乏,萎縮,毛細血管拡張症がすべて現れる)を発症する.年少期から前癌状態の日光性角化症が生じる.XPは進行性の光加齢の一例である.XP患児の日光に曝された皮膚は,長年,過度の日光にさらされてきた農夫や水夫の皮膚に類似している.
- 眼.眼の異常は皮膚の異常と同程度に多い[Kraemer et al 1987,Kraemer et al 1994].症状が10歳前に現れることもあるが,臨床症状が生じるのは紫外線の曝露を受ける前方構造(結膜,角膜,眼瞼)に限られている[Dollfus et al 2003,Brooks et al 2013].結膜に炎症の良性腫瘤が生じると,広がって,角膜混濁をきたすことがある.眼の紫外線に曝される部位に上皮腫,扁平上皮癌,黒色腫を生じることが多い.眼症状は黒人患者で重症化しやすい[Dollfus et al 2003,Ramkumar et al 2011,Brooks et al 2013].
- 神経系.106人の患者の約25 %で,緩徐進行性の神経学的異常の悪化が報告されている[Rapin et al 2000,Kraemer et al 2007,DiGiovanna & Kraemer 2012,Lai et al 2013,Totonchy et al 2013,Viana et al 2013].発症年齢は乳児初期であるが,10歳代以降になる場合もある[Rapin et al 2000].神経学的異常は軽度(孤発性の反射減弱など)の場合もあれば,小頭症,進行性認知障害,高音域から始まる感音難聴,痙直,運動失調,痙攣発作など,重度の場合もある.上気道感染が起きると嚥下困難や,稀に声帯麻痺が生じる患者もいる[Ohto et al 2004].
神経学的症状を呈する患者の剖検で見つかることの多い神経病理学的異常は,特に大脳や小脳でのニューロン喪失(もしくは欠損)である.続発性の脱髄が生じた幾つかの症例では,末梢神経の一次性軸索変性を示す所見があった[Rapin et al 2000,Lai et al 2013,Viana et al 2013].
神経伝導検査では,神経伝導速度の低下も認めることがある.
- 皮膚腫瘍.早期から積極的に紫外線遮断を行わないと,日光誘発性のDNA損傷が累積し,10歳未満で皮膚癌が生じやすくなる.Bradford et al [2011]によれば,20歳未満のXP患者では以下の癌のリスクが高くなる:
- 紫外線曝露部位の黒色腫以外の皮膚癌(基底細胞癌や扁平上皮癌).発症リスクは米国の一般人口の10,000倍超であり,発症年齢の中央値は9歳で,一般人口より約60年早い.
- 皮膚黒色腫.発症リスクは米国の一般人口の2000倍超であり,発症年齢の中央値は22歳で,一般人口より30年以上早い.
- 驚くべきことに,最重度の日光過敏症を有するXP患者の皮膚癌発症年齢は遅い.この理由として,こうした患者が行っている遮光手段が優れていることが考えられる.
他の腫瘍.全世界で報告された文献のレビューによれば,相当数のXP患者に口腔内腫瘍が生じており,特に日光曝露部位と考えられる舌先に扁平上皮癌が生じていることがわかった[Kraemer et al 1987,Kraemer et al 1994,Butt et al 2010].
脳や脊髄の神経膠腫,肺癌,子宮癌,乳癌,膵癌,胃癌,腎癌,精巣癌,白血病が少数のXP患者で報告されている[DiGiovanna et al 1998,Bradford et al 2011,Lai et al 2013,Fassihi et al 2016].
タバコの煙に含まれる発癌物質にはDNAと結合して,ヌクレオチド除去修復機能によって修復される損傷を生じさせるが,こうした未修復のDNA損傷により,XP患者が肺癌を発症することがある.総じて,こうした報告書では,XPでの内臓腫瘍は約10~20倍であると言われている[Kraemer et al 1987,Kraemer et al 1994,Bradford et al 2011].
死因.死因では皮膚癌(34 %,n=10)が最多であり,このほか,神経変性(31 %,n=9),内臓癌(17 %,n=5)がある.神経変性をきたしたXP患者の死亡年齢(中央値)は29歳であり,神経変性のないXP患者の死亡年齢(中央値)である37歳より早い(p=0.02).
遺伝子と表現型の相関
全般的な臨床的障害(XP,コケイン症候群[CS],裂毛症[TTD],脳・眼・顔・骨格[COFS]症候群など[図1を参照])に関していえば,臨床的表現型は,幅広い疾患群のなかで,変異が生じた特異的遺伝子と相関している(図1と表2を参照).
表2.
XPと関連疾患における遺伝子と表現型の相関
| 遺伝子 | 皮膚腫瘍 | 表現型 |
|---|---|---|
| DDB2 | +1 | 神経学的異常を伴わないXP |
| ERCC1 | – | COFS症候群2 |
| ERCC2 3 | + | 神経学的異常(なし~重度)を伴うXP |
| + | XP/コケイン症候群(CS) | |
| + | XP/裂毛症(TTD) | |
| - | 裂毛症(TTD) | |
| - | COFS症候群 | |
| ERCC3 4 | + | XP/コケイン症候群(CS) |
| - | 裂毛症(TTD) | |
| + | 軽度の神経学的異常を伴うXP | |
| ERCC4 | + | 神経学的異常を伴わないXP/遅発性の重度の神経学的異常を伴うXP5;ファンコニ貧血(FA)6;XP,コケイン症候群(CS),ファンコニ貧血(FA)の特徴を呈する患者が1人,コケイン症候群(CS)の特徴を呈する患者が2人7. |
| ERCC5 | + | 神経学的異常を伴わないXP,もしくは重度の神経学的異常を伴うXP |
| + | XP/CS | |
| POLH | + | 神経学的異常を伴わないXP8 |
| XPA | + | 神経学的異常(軽度から重度)を伴うXP |
| XPC | + | 神経学的異常を伴わないXP9,10 |
COFS =脳・眼・顔・骨格
XP/CS =色素性乾皮症-コケイン症候群複合型
XP/TTD =XP症状を伴う裂毛症
TTD = 裂毛症(XP症状なし)
- 多数の皮膚癌を発症した成人患者が報告されている[Oh et al 2011,Fassihi et al 2016].
- ERCC1遺伝子の両アレルで病原性多型が報告されている患者は,COFS症候群の1人の患者のみである[Jaspers et al 2007].ERCC1遺伝子にホモ接合型の病原性多型をもつ1人の患者は,重度の2型コケイン症候群を発症し,2.5歳で死亡した[Kashiyama et al 2013].
- ERCC2 (XP-D) 遺伝子の変異によってXPを発症した患者には,XP,神経学的異常を伴うXP,XP/コケイン症候群(CS)複合型,裂毛症(TTD),XP/裂毛症(TTD)[Broughton et al 2001,Lehmann 2001,Fassihi et al 2016]が生じる.
- Robbins et al [1974],Weeda et al [1997],Oh et al [2006],Fassihi et al [2016].
- 患者のほとんどが日本からの報告である.英国でも数人の報告がある[Fassihi et al 2016].
- Bogliolo et al [2013]
- Kashiyama et al [2013]
- 異型XPの患者は,臨床的には皮膚症状を有する他のXP患者と同じであるが,神経学的異常は現れない[Inui et al 2008,Fassihi et al 2016].
- 「XPでの神経学的異常」とは,ニューロン喪失に起因すると考えられる運動機能,感覚機能,認知機能の喪失の進行である.
- XPC(XP-C)遺伝子の変異によって生じるXP患者の大多数には,XPでの神経学的異常を伴わないXPが生じる[Cleaver et al 1999,Bradford et al 2011,Fassihi et al 2016].
遺伝型と臨床型の関連
遺伝型と表現型との相関に関する研究が進行中である.詳細情報は文献レビューやClinical Utility Gene Cardを参照されたい[Cleaver et al 1999,Schubert et al 2014,Fassihi et al 2016].
病名
色素性乾皮症は,モーリッツ=カポジ(Moriz Kaposi)が義父のフェルディナンド・ヘブラ(Ferdinand Hebra)とともにウィーンで1870年に出版した皮膚科学の教科書で初めて報告された.この疾患は当初,「乾皮症」や「羊皮紙様皮膚」と呼ばれた.Kraemer et al [1987]やDiGiovanna & Kraemer [2012]での議論を参照.
従来,神経学的異常を有するXP患者は,デ・サンクティス-カッチオーネ(DeSanctis-Cacchione)症候群と呼ばれていた.XPの疾患スペクトラムが明確になった現在では,デ・サンクティス-カッチオーネ症候群という病名は,低身長症と性発達遅延を呈する重度の神経症状を伴うXPにのみ用いられている.デ・サンクティス-カッチオーネ症候群の症状が全に揃う患者は非常に少数であるが,1つ以上の神経症状を呈するXP患者は多い.
今日では,「色素性類乾皮症」が異型XP(XP variant)と同一であることがわかっている.
XPの原因遺伝子が同定されるまでは,患者の機能障害を分類する際に,相補性群が用いられてきた.XP相補性試験では,患者から採取した細胞を検査室で融合して欠損が異なるかを判定する.欠損が異なる場合,細胞の正常な表現型を再建するために必要なすべての機能が提供される.このため,相補性試験は機能試験であり,患者の欠損が同一か異なるかを判断できる.その後,各相補群が別個の遺伝子の欠損により生じることがわかった(「表3」).現在,相補性群を判定する検査は市販されていない.
表3.
XP関連遺伝子と相補性群
| 遺伝子 | 相補性群1 |
|---|---|
| DDB2 > | E |
| ERCC1 | 脚注2を参照 |
| ERCC2 | D |
| ERCC3 | B |
| ERCC4 | F |
| ERCC5 | G |
| POLH | 異型XP |
| XPA | A |
| XPC | C |
- DiGiovanna & Kraemer [2012]
- これまではH群と呼ばれていたが,その後,取り消された.
遺伝的に関連がある疾患
XPの原因遺伝子の病原性多型は,本稿で扱ったXPという表現型に加え,脳・眼・顔・骨格(COFS)症候群,コケイン症候群,ファンコニ貧血(FA),裂毛症(TTD),XFE早老症候群といった表現型と関連している(図1).
表4.
原因遺伝子を同じくする疾患(Allelic Disorders)
| XP関連遺伝子 | 原因遺伝子を同じくする疾患 | 臨床徴候 |
|---|---|---|
| ERCC2 ERCC5 |
脳・眼・顔・骨格症候群(Pena-Shokeir症候群2型;OMIM) | 「鑑別診断」の項を参照. |
| ERCC1 1 ERCC4 1 |
コケイン症候群 | 「鑑別診断」の項を参照. |
| ERCC4 | ファンコニ貧血 | 身体的異常,骨髄不全,及び悪性腫瘍のリスク増加といった異なる性質をもつ疾患. |
| ERCC3 ERCC2 |
裂毛症(OMIM) | 「鑑別診断」の項を参照. |
| ERCC4 | XFE早老症候群(OMIM) | 脚注2を参照 |
- コケイン症候群の2人の患者と,コケイン症候群,色素性乾皮症,ファンコニ貧血の特徴を呈する1人の患者において,ERCC1遺伝子,もしくはERCC4遺伝子のいずれかの両アレルに病原性多型が報告されている[Kashiyama et al 2013].
- Niedernhofer et al [2006]は,悪液質,低身長症,小頭症,出生時からの顕著な日光過敏症,視神経萎縮に起因する視力障害,難聴,軽度の学習障害,進行性の成長不全,早老を特徴とする顔貌,軽度の運動失調,協調運動障害を呈する15歳男性を報告した.Niedernhofer et al [2006]は,この症例を新たな早老症候群として提唱した.XFE早老症候群が独立した疾患であるか,XP,ファンコニ貧血,XP/コケイン症候群複合型に含まれるかどうかについては確定していない.
孤発性腫瘍.癌リスクの上昇とXP原因遺伝子のヘテロ接合型のアレル多型との関連について,活発な議論が行われている.
鑑別診断
色素性乾皮症(XP),神経学的異常を伴うXP,コケイン症候群(CS),XP/CS複合型,脳・眼・顔・骨格(COFS)症候群,COFS/TTD,CS/TTD複合型,紫外線高感受性症候群[Horibata et al 2004,Berneburg & Kraemer 2007,Kraemer et al 2007,Stefanini & Kraemer 2008,Kraemer & Ruenger 2012,Ruenger et al 2012]は,ヌクレオチド除去修復(NER)障害から生じる皮膚の光過敏症を呈する10の遺伝疾患である.13の遺伝子における障害に関連している(図1を参照).
表5.
皮膚の光過敏症を呈する常染色体劣性のヌクレオチド除去修復障害
| 表現型1 | 遺伝子 | 臨床徴候 |
|---|---|---|
| 脳・眼・顔・骨格(COFS)症候群(Pena-Shokeir症候群2型;OMIM) | ERCC22 ERCC52 ERCC63 |
頭蓋内石灰化を伴う小頭症と成長障害を特徴とする進行性の精神疾患 先天性の関節拘縮とともに,小角膜,白内障,視神経萎縮といった眼症状が現れる. 細胞の紫外線過敏症と同時に光過敏症が生じることがある. |
| コケイン症候群(CS) | ERCC6 ERCC84 |
コケイン症候群(CS)1型:出生前の発育は正常であるが,2歳までに発育・発達異常が現れる.症状が完全に現れる頃には,身長,体重,頭囲が5パーセンタイルをはるかに下回るようになる.進行性の視覚障害,進行性の難聴,中枢神経系や末梢神経系の機能障害により重度の障害に至る.通常,10歳~20歳前に死亡する. XPと同様,コケイン症候群(CS)患者から採取した細胞は紫外線による細胞殺傷性への感受性が高いが,コケイン症候群(CS)細胞での紫外線照射後の不定期DNA合成(UDS)機能は正常である.コケイン症候群(CS)細胞でも転写と共役したヌクレオチド除去修復(TC-NER)が損なわれているために,紫外線曝露後のRNA合成の回復は遅れる. |
| 裂毛症(TTD;OMIM) | ERCC32 ERCC22 GTF2H5 MPLKIP5 GTF2E26 |
光線過敏症,魚鱗癬,偏光顕微鏡で「トラの尾状(tiger tail)」にみえる脆弱毛7,知的障害,低身長,小頭症,脳の髄鞘形成異常,突出した耳介と小顎症を有する特徴的な顔貌といったさまざまな表現型を伴う. 10歳以前の死亡リスクは20倍であり,主な死因は感染である8. 裂毛症(TTD)では,妊娠期の合併症と新生児期の異常の頻度が増加するが,同一遺伝子(ERCC2)に別の病原性バリアントを有するXP患者の母親では増加しない9. |
| ERCC22 ERCC32 ERCC42 ERCC510, 11 |
XP特有の顔面のそばかすと早期の皮膚癌,幾つかのコケイン症候群(CS)の特徴(知的障害,痙直,低身長,性腺機能低下).骨格異形成はなし.XP/CSでは,ニューロン変性が目立つXPとは対称的に,コケイン症候群(CS)特有の髄鞘形成異常を認める. | |
| COFS/TTD | ERCC212 | 脳・眼・顔・骨格症候群(COFS)と裂毛症(TTD)の混合型 |
| CS/TTD複合型 | ERCC212 | コケイン症候群(CS)と裂毛症(TTD)の特徴の混合型 |
| 紫外線高感受性症候群 | ERCC613 ERCC813 UVSSA13 |
色素異常や明らかな中枢神経系異常を伴わない軽度の光過敏症.患者から採取した細胞は,コケイン症候群(CS)患者でみられるのと同様の転写障害を呈する |
| XP/TTD複合型 | ERCC214 | XPの臨床的表現型と細胞表現型を呈する裂毛症(TTD)の表現型大多数の裂毛症(TTD)患者とは異なり,XP/TTD患者では皮膚癌の発症率が上昇することがある. |
MOI = 遺伝形式
AR = 常染色体劣性
- このほかの情報については,他疾患のGeneReview,OMIM,表現型,引用文献を参照.
- XPと原因遺伝子を同じくする疾患である.
- Meira et al [2000],Graham et al [2001]
- コケイン症候群の2人の患者と,コケイン症候群,色素性乾皮症,ファンコニ貧血の特徴を呈する1人の患者において,ERCC1遺伝子,もしくはERCC4遺伝子のいずれかの両アレルの病原性多型が報告されている[Kashiyama et al 2013].
- Broughton et al [2001],Itin et al [2001],Bootsma et al [2002],Giglia-Mari et al [2004],Liang et al [2005],Kraemer et al [2007],Heller et al [2015]
- Kuschal et al [2016]
- Liang et al [2005]
- Faghri et al [2008]
- Moslehi et al [2010],Tamura et al [2011],Tamura et al [2012]
- 表2と図1を参照.
- Kashiyama et al [2013]
- DiGiovanna & Kraemer [2012]を参照.
- Itoh et al [1994],Horibata et al [2004],Nardo et al [2009],Nakazawa et al [2012],Schwertman et al [2012],Zhang et al [2012],Wilson et al [2016]
- Broughton et al [2001],Boyle et al [2008],DiGiovanna & Kraemer [2012]
その他
特に他の臨床症状がない場合には,ヌクレオチド除去修復障害を呈する疾患に加え,皮膚の光過敏症を呈する疾患も検討すべきである.検討すべきこのほかの疾患には以下がある:
- ロスムンド・トムソン症候群と,原因遺伝子を同じくする疾患であるバラー・ゲロルト症候群
- ハートナップ病(OMIM)は,非極性アミノ酸輸送体をコードするSLC6A19遺伝子の両アレルの病原性多型によって生じるアミノ酸吸収障害である.ナイアシン値は低下し,ペラグラ様の光線過敏症の症状とともに皮膚炎,下痢,認知症が生じる.しかし,ハートナップ病の患者では,XP患者のように皮膚癌の発症率が上昇したとの報告はない.
カーニー複合の皮膚所見はXPと混同されることがあるが,カーニー複合の特徴は黒子であり,(多形皮膚萎縮症のような)萎縮や毛細血管拡張症といった皮膚損傷徴候が随伴することはなく,皮膚症状は日光曝露部位に限定されない[Correa et al 2015].
臨床的マネジメント
初回診断後の評価
色素性乾皮症(XP)と診断された患者の疾患の程度とニーズを判定するために推奨される評価は以下のとおり(Tamura et al [2014]のレビューを参照).
皮膚
- ベースライン時の皮膚評価(すべての日光曝露部位と遮光部位)では,日光誘発性損傷(色素性病変,前癌病変,皮膚癌など)の有無を評価する.
- 頭皮の検査では,ヘアードライヤー(送風設定)を使って髪を吹き分ける.
- 口唇や舌先付近の日光損傷徴候の有無を調べる検査では,こうした部位に癌の発症前に生じることの多い光線性口唇炎(口唇に発症する日光性角化症や白斑症の1種)や顕著な毛細血管拡張症について調べる[Butt et al 2010].
- ベースライン時に皮膚表面の全体像と個々の病変の接写像を(定規とともに)カラー撮影しておくと,経過観察や皮膚癌の早期検出に有用である.
眼
- 眼瞼と紫外線曝露を受ける眼球前方部位については,眼瞼外反,眼瞼内反,炎症性腫瘤(翼状片,瞼裂斑),角膜混濁,癌(眼瞼,結膜,角膜)といった日光誘発性の損傷の有無を調べる.粘膜表面の癌の検出するために,眼瞼を反転させなければならないことがある.
- 眼乾燥の検出にはシルマーテスト(Schirmer test)を行う.この検査では,眼瞼の下に濾紙を数分間置き,涙液吸収量を測定する[Brooks et al 2013].
神経系
- 深部腱反射検査
- 頭前後径(OFC)を測定し,小頭症であるかどうかを確認する
- 他の神経学的異常が発覚した場合には,脳MRIと神経伝導速度を検査する
聴覚
XP関連の神経学的異常の1つとして,ベースライン時のスクリーニングでの感音難聴を評価する聴力検査を行う
遺伝 臨床遺伝専門医や遺伝カウンセラーの診察
症状の治療:
症状への治療については,Tamura et al [2010b]とTamura et al [2014]のレビューがある.
皮膚.色素性病変(小さな日光性角化症など)には,液体窒素による凍結療法を行う.
これよりも大きな日光誘発性の皮膚損傷には,5-フルオロウラシル軟膏やイミキモド・クリームなどを用いた局所療法を行う.稀に,これよりも損傷の程度がひどい場合,採皮刀による薄片切除術や皮膚剥離術で表在表皮層を切除する.こうした処置を行うと,損傷部よりも紫外線曝露を受けていない濾胞細胞や腺細胞からの再増殖が可能となる.
皮膚新生物への治療は,XP患者以外の患者への治療と同じである.こうした治療には,電気乾固・掻爬術や外科的治療がある.再発した皮膚癌や,再発リスクの高い部位の皮膚癌への最適な治療はモース顕微鏡手術である.外科処置を頻回行わなくてはならない場合が多いため,損傷を受けていない皮膚の切除は最小限にすべきである.重症例への治療では,顔面表面の広い範囲を切除し,日光にさらされていない皮膚を移植する.
XP患者が治療用のX線に異常に高い感受性を示すことはなく,手術不能腫瘍に対して全照射量で治療用X線を照射したときのXP患者の奏効は,非XP患者と変わらないが[DiGiovanna et al 1998],少数のXP患者から採取した培養細胞で,X線照射への感受性が高いことがわかった[Arlett et al 2006].放射線療法の適応となる場合,治療への過敏性の有無を確かめるため,初回の照射量を低くしておくとよい.
経口イソトレチノインやアシトレチンは,多発性皮膚癌患者における新規腫瘍の発症予防に効果的である[Kraemer et al 1988].経口イソトレチノインやアシトレチンには毒性(肝毒性,催奇形性;靱帯や腱の石灰化;骨端早期閉鎖)があるため,新たな腫瘍が活発に多数生じているXP患者にのみ用いるべきである.毒性を軽減するため,イソトレチノインやアシトレチンの投与量を減らしても奏効が得られることがある.
少数の症例報告によれば,XP患者においてイミキモド・クリームを使用したところ,皮膚癌の退縮を認めたが[Giannotti et al 2003,Nagore et al 2003,Roseeuw 2003],対照試験の報告はない.
眼.眼瞼が変形した患者には,角膜の浸潤状態を保ち,機械的外傷を防ぐため,メチルセルロース点眼薬やソフトコンタクトレンズが使用されている.
角膜混濁を伴う重度の角膜炎を有する患者では,角膜移植により視力が回復した.しかし,移植片への拒絶反応予防のために行われる免疫抑制療法により,皮膚癌のリスクが上昇することがある.
眼瞼癌,結膜癌,角膜癌は,通常,外科的に治療する.
聴覚.学業に困難をきたしている感音難聴患者にとっては,補聴器がたいへん有用である(Totonchy et al [2013],「難聴・遺伝性難聴概説」を参照).
一次病変の予防:
XPの治療では,早期診断と,診断後すぐに日光と紫外線曝露を積極的に回避する措置を開始することがきわめて重要である.こうした措置には,紫外線があたるとき(太陽が出ている時間や,曇りの昼間),屋外での曝露を避けるか,最小限に抑えることが含まれている.
XPが臨床的に疑われる場合には,診断が確定されたり,他の原因が判明することを待たず,すぐに日光防護措置をとるべきである.
患者は帽子,長袖,長ズボン,手袋を着用したり,SPF(日焼け防止指数)の高い日焼け止めを使用したり,紫外線を吸収する眼鏡をつけたり,髪を長く伸ばしたりするなどして,全身の表面が紫外線に当たらないようにするように指導を受けるべきである.眼については,両側に覆いのついた紫外線を吸収する眼鏡を着用すること.顔面に紫外線が当たらないようにしつつも,戸外での視界を維持できるような,紫外線を吸収する顔面シールドのついた自作の帽子を着用する患者もいる.
XP患者の細胞は(日光中の)紫外線Aや紫外線B,(人工光源中に含まれる場合のある)紫外線Cに対して過敏なため,患者の自宅,学校,職場環境の紫外線量を測光露出計で測定して環境紫外線量(ハロゲンランプなど)の高い場所を確認し,可能ならば紫外線量をゼロにできるとよい.XP患者にとって絶対に安全といえる紫外線曝露量の基準はないが,紫外線測定機器を使用すると,予期せず環境紫外線が高い場所であることがわかった場合に患者自身が警戒できる.
二次合併症の予防:
ビタミンDは紫外線への曝露が関与する反応により,皮膚で産生される.皮膚癌を有する活動的な成人XP患者では,過去の食事で十分量のビタミンDを摂取しているため,活性型(1,25-ジヒドロキシビタミンD)の血清濃度は正常である[Sollitto et al 1997].しかし,生後きわめて早くから遮光対策を始めた患児の25-ジヒドロキシビタミンD血清濃度は低く,1人の患児では骨折が起きやすくなった[Ali et al 2009;著者の個人的観察所見].ビタミンDの血清濃度が低い患者に対しては,ビタミンDの経口サプリメントの摂取が推奨される[著者の個人的観察所見].
経過観察:
Tamura et al [2010b]とTamura et al [2014]を参照.
皮膚.医師は患者の皮膚を定期的に診察すること(皮膚病変の重症度に応じて3~12ヶ月に1回).
患者や患者の親は,異常な色素沈着病変や,基底細胞癌や扁平上皮癌のようにみえる病変の発見方法について,指導を受けるべきである患者は皮膚癌の発見方法を指導された家族に,頻繁に皮膚を見てもらうとよい.
眼.紫外線曝露徴候や損傷がないか,定期的に眼検査を行うこと.
神経系.進行性の神経学的異常を認めるXP患者が少数ながら存在するが,年少児ではみつけられないこともあるため,定期的に神経学的検査と聴力検査を行うとよい.
聴覚.定期的に聴力検査を行う.特にごくわずかな日光曝露で急性熱傷が生じたことのある患者では,一定の間隔をおいて連続して聴力図を検査すると,進行性の神経学的悪化の有無を評価する上で有用であろう.
回避すべき薬剤・環境:
日光や紫外線の人工光源への曝露は避けるべきである(「一次病変の予防」を参照).
紫外線の人工光源.ある種の光源(水銀灯,ハロゲンランプ,その他のランプなど)が紫外線を生じさせることはあまり知られていない.こうした光源に遮光措置がとられていることが多いが,体育館のような広い場所では,遮光物に隙間があると紫外線曝露の原因となりうる.予測できない紫外線発生源を特定し,周囲の状態を容易に監視することのできる紫外線測定器はすぐに入手できる.
タバコの喫煙.XP患者の細胞はタバコの煙に含まれるベンゾ[ア]ピレンなどの環境変異原への過敏性も高いため,XP患者はこうした物質からも慎重に身を守るべきである.喫煙歴が10年超のXP患者の1人が,35歳に気管支原性肺癌で死亡した[Kraemer et al 1994].近年,著者は40歳代に肺癌を発症した喫煙歴のあるXP患者を診察した.
リスクのある近親者の検査:
できるだけ早期に治療を開始したり,予防措置を講じたりすることが重要であるため,一見,無症状の発端者の兄姉や弟妹に対して検査を行うことは妥当な措置である.
- 家系特異的な病原性多型が確認されたが,リスクのある同胞に対して分子遺伝学的検査を行うことが可能となる.
- 発端者の同胞が罹患しているかどうかを確認する臨床的評価は,同胞が乳児や幼児である場合,困難となることがある.こうした場合には,臨床検査が確実に行うことができるようになるまで,同胞に対して日光防護を行うことが推奨される.
遺伝カウンセリングを目的としたリスクのある近親者の検査に関連する問題については,「遺伝カウンセリング」の項を参照.
妊娠管理
新たな腫瘍が活発に多数生じているXP患者には,皮膚癌の化学予防薬として,イソトレチノインやアシトレチンによるレチノイドの全身投与が行われる.このため,XPの女性患者に対してこうした薬剤が使用されることがある[Kraemer et al 1988].レチノイドの全身投与には発育中の胎児への催奇形性があり,胎児の先天性欠損のリスクを高めることがわかっている.このため,レチノイドの全身投与を受けている女性は,妊娠のリスクと効果的な避妊の必要性について適切なカウンセリングを受けるとよい.また,定期的に妊娠検査を行ってモニタリングをすべきである.同薬によるスクとベネフィットを熟知した医師のみが,レチノイドの全身投与を行うこと.
米国では,女性患者や女性患者への処方者がイソトレチノインを入手するためには,胎児曝露の可能性を最小限にするため,iPLEDGEプログラムに登録しなければならない.治療前から避妊を開始し,投与中も継続し,投与終了後も薬剤が完全に身体から排出されるまで避妊を継続する.どちらの薬剤も皮膚癌の予防には有効であるが,アシトレチンの方が体内からの排出が遅く,催奇形リスクを最小限にするために避妊を継続すべき期間が長い(3年).
研究中の治療
細菌性DNA修復酵素であるT4エンドヌクレアーゼVを含むリポソーム含有局所製剤が,XP患者での日光性角化症と基底細胞癌の新規発症率を低下させたとことが,1件の臨床研究で報告された[Yarosh et al 2001].2016年現在,この治療法は米国食品医薬品局(FDA)の承認を受けていない.
遠隔転移を伴う基底細胞癌や手術後に再発をきたした局所進行基底細胞癌の治療薬として,ヘッジホッグ経路阻害薬であるビスモデギブ(エリベッジ)(vismodegib[Erivedge®])がFDAの承認を受けた.同薬は,手術,もしくは放射線療法が適応されない基底細胞癌治療薬としても承認を受けている(FDA添付文書を参照).同薬の適応となるXP患者もいるが,XP患者における同薬の有効性に関する研究は発表されていない.ビスモデギブ経口薬にも催奇形性があり,胎芽-胎児死亡や,胎芽や胎児の正中線異常(midline defects),指喪失,他の先天性欠損を生じさせる.ビスモデギブ投与中と投与後は,男女ともに有効な避妊が指示される.
種々の疾患の臨床試験に関する情報については,こちらを参照されたい.
その他.
悪性細胞の細胞学的検査のため,結膜病変からの検体を採取する際に綿棒を用いることに関して,評価が行われている[Brooks et al 2013].
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
色素性乾皮症(XP)の遺伝形式は常染色体劣性である.
患者家族のリスク
発端者の親
- XPの子をもつ両親は絶対的ヘテロ接合体(XP関連遺伝子の病原性多型の保因者)である.
- XPに関連する病原性多型のヘテロ接合体(保因者)に臨床症状は現れない.
発端者の同胞
- 受精時に同胞が発症する確率は25 %,無症候性の保因者となる確率は50 %,発症せず,保因者ともならない確率は25 %である.
- ヘテロ接合体(保因者)に臨床症状は現れない.
発端者の子
- XP患者の子はXP関連の病原性多型の絶対的ヘテロ接合体(保因者)であり,臨床症状が現れることはない.
- XP患者や,臨床的に無症状のXP関連の病原性多型のヘテロ接合体の子がXPを発症する確率は50 %である.これは創始者変異を有する集団や同族結婚率が高い集団において考慮される.
発端者の他の血縁者
-
発端者の両親から生まれた同胞がXP関連の病原性バリアントの保因者であるリスクは50 %である.
保因者(ヘテロ接合体)診断
リスクのある血縁者への保因者診断に際しては,家系内でXP関連の病原性多型が同定されていなければならない(Christen-Zaech et al [2009]の議論を参照).
保因者であることがわかっている配偶者への保因者診断は,幾つかのXP関連遺伝子多型については可能である.
遺伝カウンセリングに関連した問題
リスクのある血縁者への早期診断・治療目的の評価に関する情報については,「臨床的マネジメント」,「リスクのある近親者の検査」の項を参照されたい.
家族計画
- 遺伝リスクの判定,保因者胴体の確認,出生前診断を利用するかどうかに関する議論を行う最適な時期は妊娠前である.
- 罹患者であったり,保因者であったり,保因者リスクがあったりする若年青年に,(子へのリスクや生殖における選択肢などの議論も含めた)遺伝カウンセリングを提供することは妥当な措置である.
DNAバンキングは,将来の使用のために,通常は白血球から調整したDNAを貯蔵しておくことである.検査手法や,遺伝子,変異,疾患への理解は将来改善する可能性があり,罹患者のDNAを貯蔵しておくことは考慮されるべきである.
出生前診断と着床前診断
罹患者家系でXP関連遺伝子の病原性変異が同定されれば,XPのリスクの高い妊娠に対しての出生前診断や着床前遺伝子診断を行うことも可能である.
更新履歴
- Gene Review著者: Kenneth H Kraemer, MD, John D DiGiovanna, MD
日本語訳者: 窪田美穂(ボランティア翻訳者),奥山隆平(信州大学医学部皮膚科学講座)
Gene Review 最終更新日: 2012.3.15. 日本語訳最終更新日: 2012.5.1. - Gene Review著者: Kenneth H Kraemer, MD,John J DiGiovanna, MD.
日本語訳者: 窪田美穂(ボランティア翻訳者),櫻井晃洋(札幌医科大学医学部遺伝医学)
Gene Review 最終更新日: 2014.2.13. 日本語訳最終更新日: 2014.9.5 - Kenneth H Kraemer, MD and John J DiGiovanna, MD.
日本語訳者: 窪田美穂(ボランティア翻訳者),櫻井晃洋(札幌医科大学医学部遺伝医学)
Gene Review 最終更新日:2016.9.29,日本語訳最終更新日:2017.1.15 (in present)