これまでの研究成果(KLF5)
①心血管病におけるKLF5
私たちは血管平滑筋細胞が血管病変を形作るのに重要なことに着目し、平滑筋細胞の機能を司る分子メカニズムの研究を続けてきました。平滑筋細胞は、骨格筋細胞や心筋細胞とは異なり、いったん高度に分化した後も、様々な刺激に応じて性質(形質)を変えます。これを形質変換(phenotypic modulation)といいますが、血管病のでき方を理解するためには平滑筋細胞の形質変換、また形質変換とは裏腹の変化である分化の分子メカニズムを知ることが大事です。
__平滑筋形質変換とKLFファミリー転写因子__
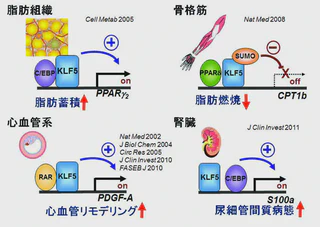
KLF5の機能をさらに明らかにするために、ノックアウトマウスを作りました(Nat Med. 2002;8:856-863)。一対のKLF5遺伝子の両方を欠いたホモ接合体ノックアウトマウスは胎児期に死んでしまうため、2本のKLF5遺伝子のうちの一つが残っているヘテロ接合体ノックアウトマウスを用いて解析を行いました。何もしない状態ではヘテロ接合体ノックアウトマウスの血管に著明な異常は認められませんでしたが、いったん血管に傷をつけると、その結果生じる病変(新生内膜)がほとんどできないことが分かりました。このことから、KLF5が傷害に対する血管の反応と病変形成に必須な鍵分子であることが分かります。KLF5は血管の組織構築を改変するのに重要なPDGF-Aなどのパラクライン因子の発現を制御するとともに、平滑筋形質変換を制御して病変形成を調節していると考えられます(Circ Res. 2005;97:1132-1141)。KLF5ヘテロ接合体ノックアウトマウスでは、各種の負荷による心臓の肥大や線維化も減弱しており、血管だけでなく心臓の組織構築の改変(リモデリング)に重要であることが明らかとなっています。
__TACによる心肥大__

__心筋細胞Klf5欠損__

__心筋を構成する細胞__

__線維芽細胞Klf5欠損は心肥大を抑制する__
②代謝疾患におけるKLF5
面白いことにKLF5ノックアウトマウスでは生後しばらく皮下脂肪 (白色脂肪組織)が薄く発達が悪い。この観察から、KLF5が脂肪細胞の分化を制御する転写因子ネットワークの重要な構成要素であることを見つけ出しました(Cell Metab. 2005;1:27-39)。心血管系の病気で重要な鍵分子として見つけ出したKLF5が代謝組織でも重要なことは驚きでした。私たちは、KLF5がさらに成体でも代謝ストレスに対する応答に重要であることを見いだしています。第3項でみるように肥満やメタボリックシンドロームを背景とする血管と代謝組織の変化には多くの共通点があります。私たちは、KLF5が血管と代謝組織の両方で機能して、メタボリックシンドロームにおいて血管と代謝組織の両方で同時に病気を進める重要な因子であると考えています。
__血管と脂肪組織のKLF5__

__KLF5は骨格筋脂肪燃焼の分子スイッチである__
③慢性腎臓病におけるKLF5
慢性腎臓病(CKD)は、透析が必要な慢性腎不全の原因となるだけでなく、心不全や脳梗塞などの様々な生活習慣病のリスクとなるとともに、予後に大きく影響することが分かってきています。そのため、CKDのメカニズムを明らかにし、新たな予防法・治療法を開発することが強く求められています。疾病構造の変化に伴い、最近では透析の原因となる原疾患は、糖尿病や高血圧が増加しています。このようなCKD の原因疾患の変化とともに、その発症機序については慢性炎症が注目されるようになっています。尿細管間質領域の障害がfinal common pathwayとしてCKDの予後を規定する最重要な要因であることが知られていますが、尿細管間質領域の障害を生じる主要なメカニズムが慢性炎症で、線維化や尿細管の破壊などの組織構築のリモデリングを引き起こします
KLF5はマウスの腎臓では集合管上皮細胞だけに特異的に発現が認められました。KLF5ヘテロノックアウトマウス(Klf5+/-)にCKDのモデルである片側尿管結紮術(UUO)を施したところ、Klf5+/-で組織の破壊が著明に抑制されていました。ところが、線維化はなぜかむしろ増悪することが分かりました。
__KLF5欠損による腎組織障害抑制__

__集合管上皮細胞のKLF5は初期炎症を制御する__