TDP-43 proteinopathy
神経変性疾患とは、神経細胞の構造や機能が徐々に失われていくことを指す言葉で、筋萎縮性側索硬化症(ALS)、前頭側頭葉変性症(FTLD)、アルツハイマー病(AD)、パーキンソン病(PD)、ハンチントン病(HD)など、多くの神経変性疾患の特徴である。神経変性疾患の予防法や、改善させる治療法はない。これらの疾患における神経変性の原因はほとんど解明されていないが、ごく一部の疾患では、種々のタンパク質の遺伝子変異と関連している。過去10年の間に、ALS、FTLD、ADなどの神経変性疾患には、TAR DNA‐binding protein 43 (TDP-43) proteinopathyとして知られる共通の病理学的特徴があることが明らかになってきている。神経毒性の引き金としてのTDP-43の役割は、in vitroおよびin vivoのさまざまな実験モデルで報告されている。ここでは、様々な主要な神経変性疾患における疾患進行過程におけるTDP-43 proteinopathyについて考察した後、特にALS、FTLD、ADに焦点を当てて、潜在的な病態メカニズムに関するこれまでの研究と最新の研究をレビューし、神経変性疾患に対する治療法としてTDP-43を標的とした治療法の可能性について述べる。
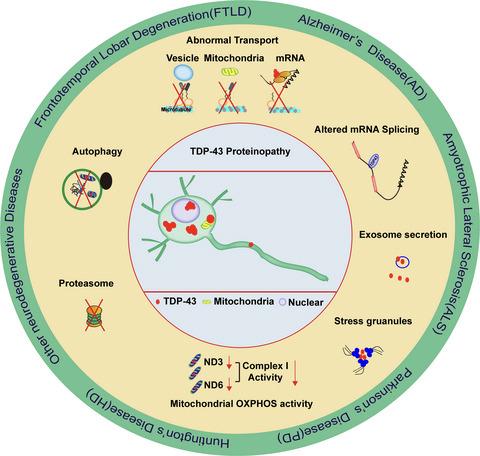
神経変性疾患は、加齢に伴う中枢神経系(CNS)または末梢神経系(PNS)のニューロンの進行性の損失または機能不全を特徴とする多数の疾患を含む。アルツハイマー病(AD)、パーキンソン病(PD)、ハンチントン病(HD)、筋萎縮性側索硬化症(ALS)および前頭側頭葉変性症(FTLD)は、よく知られている神経変性疾患である。神経変性疾患は、進行性の神経細胞障害が共通の特徴であるが、特定の部位のCNSに神経細胞消失があり、病理学的・臨床的に特徴のある疾患である。神経変性疾患の有病率は、過去数十年の間に急速に増加している。しかし、これらの疾患に対する効果的な治療法はまだ非常に限られている。
TAR DNA‐binding protein 43(TDP-43、TARDBP遺伝子によってコードされ、ALS10とも呼ばれる)は、ヒト免疫不全ウイルス1型のTARとして知られる長い末端リピートのピリミジンに富んだDNAモチーフに特異的に結合し、ウイルス1型遺伝子の転写を抑制する宿主細胞タンパク質として最初に同定された。しかしながら、その後の研究により、TDP-43はRNAとより頻繁に結合し、メッセンジャーリボ核酸(mRNA)のスプライシング、翻訳、輸送、さらには分解を調節することが実証されている。TDP-43と神経変性疾患との関連は、ALSおよびFTLD患者における病理学的特徴であるユビキチン陽性タンパク質内包物の主要構成要素としてのTDP-43の最初の同定によって確立された後、直ぐに、ALSとFTLDの両方に関連するTDP-43の遺伝的変異が発見された。それ以来、TDP-43は神経変性疾患の分野で注目されている。
病因、臨床症状、および病理学的特徴が異なるにもかかわらず、様々の神経変性疾患は、神経細胞やグリア細胞において同様のTDP-43病理学的特徴、すなわち、核内のTDP-43減少を伴う、細胞質内の界面活性剤耐性で、ユビキチン化またはリン酸化TDP-43封入体の蓄積が観察される。これらの特徴的なTDP-43関連の病理学的特徴は、TDP-43 proteinopathyと呼ばれている。ALS、FTLD、AD、PD、およびHDを含む種々の神経変性疾患における共通の重要な病理学的特徴としてのTDP-43蛋白質障害の存在は、TDP-43プロテインオパシーの病態メカニズムへの関心を、過去10年間、大幅に高めている理由である。このレビューでは、まず、ALS、FTLD、およびADに特に重点を置いて、様々な主要な神経変性疾患におけるTDP-43 proteinopathyの病期および進行について説明し、TDP-43の潜在的な病態メカニズムについて広範に議論し、最後に、神経変性疾患を治療するための一般的な治療アプローチとしてTDP-43を標的とすることの可能性について述べる。
Stages of TDP‐43 proteinopathy in ALS, FTLD, and AD
Amyotrophic lateral sclerosis
ALSは、ルー・ゲーリック病としても知られている最も一般的な運動ニューロン疾患であり、上位下位の運動ニューロンが進行性で致死的に変性し、筋肉の変性、脱力、萎縮、痙縮、麻痺、そして最終的には死に至ることを特徴とする。ALSのほとんどは家族歴がなく、通常は孤発性ALS(sALS)と呼ばれている。ALS症例の10%未満が家族性(fALS)であり、そのうち約40%がC9ORF72遺伝子のリピート延長、20%が銅-copper–zinc superoxide dismutase(SOD1)をコードする遺伝子の変異、4%がTDP-43またはfused in sarcoma(FUS)の変異によるものである。他に、p62 (SQSTM1)、optineurin, TANK‐binding kinase 1, ubiliquin‐2 (UBQLN2), vesicle‐associated membrane protein‐associated protein B, senataxin (SETX), angiogenin (ANG), valosin‐containing protein (VCP), SIGMAR1, or dynactin (DCTN1)をコードする他の多くの遺伝子の突然変異(各1%未満)がある。
sALSおよびfALSのごく一部にTDP-43変異が認められるが、TDP‐43 proteinopathyはALS患者の97%に認められることから、TDP-43はALSの病態形成に重要な役割を果たしている。神経細胞におけるTDP-43蛋白質障害に基づいて、ALSは、運動野、脳幹、脊髄に病変が出現する第1期、前頭前野、脳幹、前小脳核(橋核・橋被蓋網様核・オリーブ核・外側網様核・傍正中網様核・舌下神経周囲核:舌下神経前置核・介在核・Roller核)、赤核に病変が増加する第2期、前頭前野、中心後回、線条体、側頭葉前内側に病変が広がる第3期と、ALSの病期を分けることができる。 TDP‐43 proteinopathyの運動ニューロンの変性における役割については、まだ議論の余地がある。特筆すべきは、TDP‐43 proteinopathy はALS患者のアストロサイトやミクログリアなどのニューロン以外の細胞でも認められていることであり、TDP‐43 proteinopathy が細胞自律性と非細胞自律性(周りの環境の関与)の両方で運動ニューロン死の引き金となる可能性を示している。
Frontotemporal lobar degeneration
FTLDは、脳の前頭葉と吻側の側頭葉の変性に伴う行動や言語の進行性の低下を特徴とし、65歳以下の人ではADに次いで2番目に頻度の高い認知症である。FTLDの最も一般的な臨床症状は、典型的には、行動障害型前頭側頭型認知症 (behavioral variant frontotemporal dementia)、意味性原発性進行性認知症 (semantic variant primary progressive aphasia)、非流暢性原発性進行性認知症 (nonfluent variant primary progressive aphasia)が含まれており、行動障害型前頭側頭型認知症が最も一般的な形態として、FTLD患者の約60%を占めている。臨床的に、FTLDは進行性核上性麻痺(PSP)や大脳皮質基底核症候群(CBS)を含むこともあり、神経病理学的にもALSや他の運動ニューロン疾患(MND)と重なることがある。FTLD症例の約30~50%は家族性であり、C9ORF72、プログラヌリン(GRN)、および微小管関連タンパク質タウの遺伝的変異と一般的に関連し、まれにVCPおよびChromatin‐modifying protein 2B, TDP‐43, FUS, p62/SQSTM1, および ubiquilin 2 (UBQLN2)と関連している。C9orf72のノンコーディングG4C2ヘキサヌクレオチドリピート延長は、FTLDとALSを併発するFTLD-ALSにおいて最も一般的な原因変異として同定されている(注目すべきは、C9ORF72とは異なり、TDP-43やFUSなどの他のALS関連タンパク質の変異は、FTLD-ALSには関連しているが、MNDを欠くFTLDには関連していない)。
歴史的には、FTLD の多くは、ニューロンやグリア細胞で、封入体と呼ばれる細胞質内にタンパク質の凝集体が認められる。神経病理学的観点から、FTLDは、タウ陽性封入体を有するFTLD(FTLD-tau)、タウやα-シヌクレインは陰性であるがTDP-43およびユビキチン陽性封入体を有するFTLD(FTLD-TDP、以前はユビキチン陽性封入体を伴うFTLD、FTLD-Uと呼ばれていた)、FUS陽性介在物を伴うFTLD(FTLD-FUS)、および封入体を伴わない稀なFTLD(FTLD-niまたは特徴的な病理組織学を欠く既知の認知症と呼ばれていた)などの異なる病理学的FTLDサブタイプに分類することができる)。 FTLDにおけるTDP-43変異の有病率は非常にまれであるが、FTLD-TDPは最も頻度の高いFTLDサブタイプであり、FTLD症例の最大50%にTDP-43蛋白症(大部分がTDP-43陽性の細胞質内封入体と変性神経突起、時折TDP-43陽性の神経核内封入体)が認められる。FTLDでは、TDP-43陽性封入体は主に前頭側頭前野や海馬の歯状回に認められ、脳幹の脳神経核や脊髄前角にも認められる。
Alzheimer's disease
ADは、記憶、学習、意識的思考、言語に重要な脳領域のニューロンの消失が進行することを特徴とする高齢者の最も一般的な認知症であり、通常、2つの病理学的特徴である神経原線維変化(NFTs)と老人斑(SPs)、および神経細胞の消失、顆粒空胞変性、ジストロフィー性神経突起などの神経病理学的変化を伴っている。NFTsはリン酸化タウからなる細胞内病変であり、SPsはアミロイドβ(Aβ)ペプチド線維の束からなる細胞外病変である。最近では、細胞質TDP-43陽性封入体が、ADと診断された人の脳内で3番目に認められる蛋白質障害として注目されている。AD症例の約90%は孤発性ADと呼ばれ、遺伝的には伝播していない。AD症例の10%未満は家族性ADであり、βアミロイド前駆体タンパク質(APP)、プレセニリン1(PS1)、プレセニリン2(PS2)の変異を伴い、PS1/2は早期発症の家族性ADの大部分と関連している。
C末端TDP-43抗体を用いて検出されたTDP-43陽性封入体は、ADにおける最新の6つのステージごとに現れる。ニューロンにおけるTDP-43の細胞質蓄積は、第1期の扁桃体から始まり、第2期の嗅内野と支脚、第3期の海馬歯状回と後頭側頭回へと広がる。第4期では島回、腹側線条体、大脳基底核、下側頭回へ、第5期では黒質、下オリーブ核、中脳視蓋へ、そして最終的には第6期では大脳基底核と中前頭回へと広がる。また、最近の報告では、細胞質TDP-43封入体が病理学的特徴であるNFTやSPとは独立してAD型認知症と関連していることや、TDP-43、Aβ、タウ蛋白質障害が混在する患者は、Aβやタウ蛋白質障害のみの患者よりも重度のAD型認知症を示すことも注目に値する。
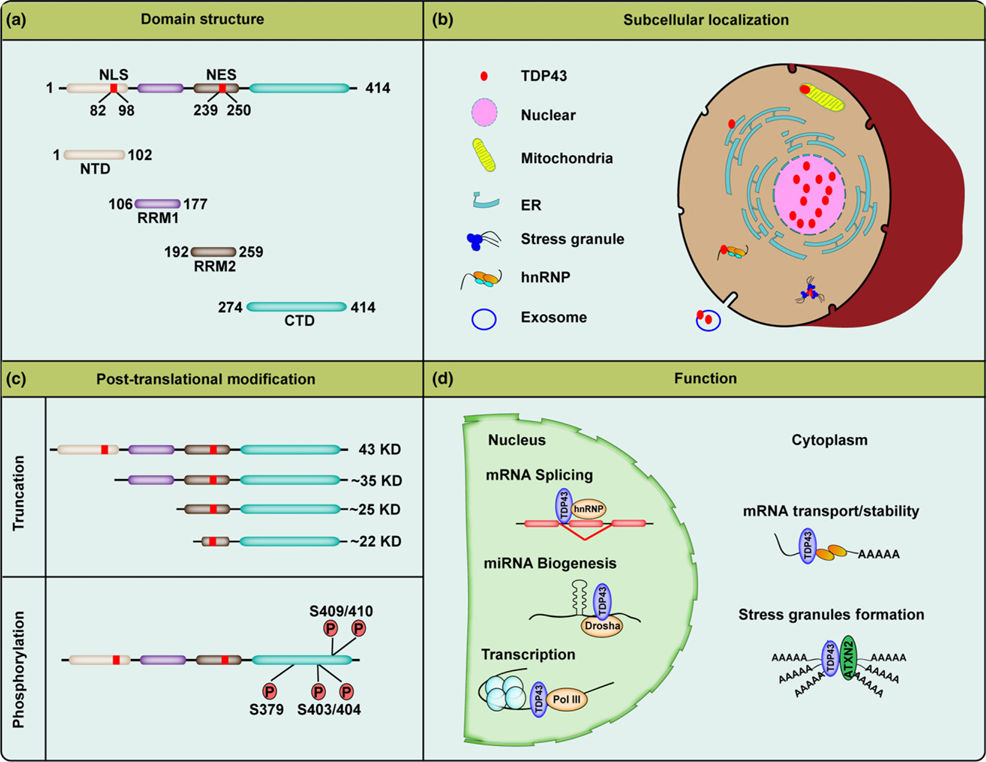
図 TDP-43のドメイン構造、細胞内局在、翻訳後修飾、および機能。
(a) TDP-43は414アミノ酸のタンパク質で、N末端ドメイン(NTD、1-102aa)、2つのRNA認識モチーフ(RRM1、106-177aaおよびRRM2、192-259aa)、およびカルボキシ末端グリシンリッチドメイン(CTD、274-414aa)を含む。TDP-43は、核局在化配列(NLS、82-98aa)と核輸出配列(NES、239-250aa)の両方を含む。
(b) TDP-43の大部分は生理的条件下では核内に存在し、残りのTDP-43はミトコンドリア、小胞体(ER)、エクソソソームなどの他の小器官に存在することがわかっている。ストレス環境下では、TDP-43はストレス顆粒やhnRNPにリクルートされる。
(c) 全長43kDaのTDP-43は、疾患条件下で、カスパーゼまたはカルパインによって切断され、~35kDaおよび20~25kDaのフラグメントを生成する。このフラグメントと全長タンパク質は、神経変性疾患に罹患した脳では、セリン残基379、403/404、409/410で異常にリン酸化される。
(d) TDP-43は複数の正常な生物学的機能を有しており、主に核内でのmRNAスプライシング、miRNAの生合成、転写などのRNA経路を制御する機能を有しているが、細胞質内でのmRNA輸送、mRNAの安定性、ストレス顆粒形成にも関与していると考えられている。
Pathomechanisms of TDP‐43
RNA alternate splicing
RNA結合タンパク質のheterogeneous nuclear ribonucleoprotein (hnRNPs) のメンバーに構造的に類似する414アミノ酸(aa)からなるTDP-43は、N末端ドメイン(NTD、1-102aa)、2つのRNA認識モチーフ(RRM1、106-177aaおよびRRM2、192-259aa)、およびC末端グリシンリッチドメイン(CTD、274-414aa)から構成されている。RRMM1およびRRMM2は、広く存在する真核生物のRNA認識モチーフファミリー(RRMMは、RNA結合ドメインまたはリボヌクレオタンパク質ドメイン(RNP)とも呼ばれる)に属する(図1a)。hnRNPsと同様に、主に核内RNA結合タンパク質として、TDP-43は、cystic fibrosis transmembrane conductance regulator, POLDIP3, SORT1, MADD/STAG2/FNIP1/BRD8, SEPT6/SULT4A1/TNIK/DICER/ELAVL3, 或いはmRNAオルタナティブスプライシングを調節するためのTARDBP自身の転写物などのmRNAの3′ 末非転写領域或いは近位イントロン部にあるUG‐rich ドメインに結合する。TDP-43欠損は、塩基配列が似ている部位をスプライス部位と誤認し異所性のスプライスサイト(cryptic splice site)を使用を促進し、本来とは異なった構造のmRNAを造り、正しくスプライスされたタンパク質をコードするmRNAを減少させ得る。TDP-43の様々なRNAへの結合のグローバルな機能は、ほとんど不明のままであるが、TDP-43が異所性のスプライシングを抑制するという知見は、TDP-43の主要な機能的RNAターゲットの同定につながる。
TDP-43 proteinopathyの顕著な特徴である、TDP-43の核から細胞質への再分配は、一般に、核機能の損失およびそれに続く神経細胞の機能障害を引き起こすと考えられている。機能喪失仮説の裏付けとして、TDP-43陽性封入体を有するALSにおいて、有意に増強されたTDP-43関連のcryptic splice siteが報告された。そして、野生型TDP-43とは異なり、外因性に発現したALS関連変異TDP-43は、神経細胞の伝達および機能に関与するmRNA標的のエクソン排除を増加させ、変異TDP-43タンパク質がmRNAスプライシング能力を失わせる。TDP-43は、FTLD関連FUS、プログラヌリン、またはタウのmRNA前駆体のスプライシングを調節し、FTLD-ALS患者は、FUSの発現が有意に減少し、プログランヌリンの発現がmRNAおよびタンパク質レベルで増加する。ごく最近の研究では、TDP-43がその3′-UTRに結合することによってタウの発現を抑制し、ALSおよび変異TDP-43を発現するALSモデル動物においてタウのタンパク質レベルが増加することが示されている。タウタンパク質発現の減少は、少なくともFTLD患者のサブセット、特にプログラヌリン変異を有するFTLD患者と関連していることが報告されている。以前はAD患者におけるTDP-43 proteinopathyとタウの発現および誤ったスプライシングとの間の関連性はないとされていたが、最近の研究では、細胞および動物モデルにおいて、TDP-43によるタウmRNA安定性およびエクソン10選択的スプライシングを調節していることが示されている。さらに、TDP-43の核発現低下を示すが、細胞質内にTDP-43陽性封入体のないADにおいて、クリプティックエクソンの取込みが起こり得ることが明らかにされている。これらの証拠は、神経変性におけるTDP-43関連RNAスプライシングの関与の可能性を支持するものであるが、機能喪失仮説は、cystic fibrosis transmembrane conductance regulatorのエクソン9の選択的スプライシングが変化していないこと、およびALS、FTLD、またはAD患者やトランスジェニック動物モデルでは他の標的mRNAが変化していないことから、疑問視されている。また、TDP-43の一般的なRNA前駆体のスプライシング活性がどのように疾患の進行に寄与しているか、また、TDP-43が多様なmRNAの選択的スプライシングを妨害して分子経路を混乱させているかについては、まだ明らかにされていない。
Stress granules
TDP-43は、核局在化配列と核輸出配列(NES)の両方を含み、それぞれNTDドメインとRRM2ドメインに局在している(図1a)。生理的条件下では、TDP-43の大部分は核内に存在する。しかし、NESが存在するためか、TDP-43の30%までが細胞質にも存在することが明らかになった(図1b)。TDP-43は核内の減少に伴い、ALS、FTLD、AD患者の神経細胞では特徴的な細胞質蓄積を示す。ALS関連TDP-43変異では、核内の減少は神経細胞毒性には必要なく、細胞質内にあるTDP-43変異が神経変性を引き起こす。核機能の喪失に加えて、TDP-43は細胞質における神経細胞毒性機能の獲得によって疾患の進行に寄与していることが示唆されている。
ストレス顆粒(SG)は、ストレスに応答して非必須タンパク質の翻訳を抑制し、保護タンパク質の発現を促進するために形成された細胞質のフォーカスであり、非膜結合型RNA顆粒として、非翻訳mRNA、翻訳開始因子(eIF2α、eIF3、eIF4A/B/G)、特徴的なマーカーを含む。非膜結合RNA顆粒として、SGsは非翻訳mRNA、翻訳開始因子(eIF2α、eIF3、eIF4A/B/G)、およびT細胞制限細胞内抗原-1(TIA-1)、TIA-like-1(TIAR)、Tristetraprolin、ポリ(A)結合タンパク質(PABP)、およびRas-GTPase活性化タンパク質SH3ドメイン結合タンパク質1などの特徴的なマーカーを含んでいる。SGに加えて、翻訳に関与していないmRNAは、RNA結合タンパク質と共集合して、processing bodies(P-body)やRNA輸送顆粒などの他の細胞質mRNP顆粒を形成する。P-bodyは、RNAのサイレンシングおよび分解を調節するためのデキャッピング酵素およびデキャッピングアクチベーターで構成され、一方、RNA輸送顆粒は、翻訳が抑制されたmRNAの分泌を担当している。
ALSまたはFTLD患者において、TDP-43またはリン酸化TDP-43は、SGメーカーであるTIA-1/PABP-1/eIF3と共局在化しており(図1b)、SGはTDP-43封入体の形成に重要な役割を果たしている。転写インヒビターを用いたSG形成の阻害により、TDP-43封入体形成の抑制はこれを支持する。また、SG形成に必要なポリグルタミン蛋白質であるAtaxin-2の機能障害は、TDP-43 proteinopathyと神経毒性を減少させる。さらに、ALSに関連するTIA-1変異は、SGの分解を遅らせ、TDP-43を含む非動的SGの蓄積を増強する。一方、TDP-43は、必須のSG成分ではないが、ストレスに応答してSGの形成および維持にも寄与している。注目すべきは、疾患関連TDP-43変異によってSG形成が増強されたことが報告されたが、他の研究では、TDP-43変異を有する細胞またはヒト初代線維芽細胞においてSG形成が減少したか、または変化しなかった。そして驚くべきことに、TIA1変異を有するALS/FTDDにおいて広範なTDP-43病理が存在するにもかかわらず、TDP-43封入体はTIA1と共局在化していない。これらの矛盾はまだ解決されていないが、ストレス応答における転写コントロールにおけるTDP-43の機能的役割はますます重要視されている。誤って局在化した細胞質TDP-43がSGのダイナミクスに影響を与え、おそらく他のRNA顆粒経路に影響を与える詳細なメカニズムを調べるためには、さらなる研究が重要である。
Axonal transport
軸索輸送は、神経細胞の機能および生存に不可欠である。軸索に沿った小胞体またはミトコンドリア輸送の異常は、ALS患者およびAD患者において広く報告されている。別のALS原因タンパク質FUSと同様に、ALS関連TDP-43変異は、mRNAを含むTDP-43顆粒の微小管依存性軸索輸送を抑制する。神経細胞の樹状突起には、TDP-43に富み、局在的翻訳を調節している。 また、TDP-43は、VEGFA/GRN, survival motor neuron (SMN) protein/fragile X mental retardation protein (FMRP), neurofilament light chain (NFL), actin/CAMKII, やMTHFSD/DDX58などをコードするmRNAを含む顆粒を形成し、mRNAの分解、輸送、および翻訳を調整している(図1d)。野生型または変異型TDP-43の過剰発現はミトコンドリア輸送を障害し、mRNAやミトコンドリア、および局在的転写や代謝に関連するタンパク質の輸送を障害する細胞質内TDP-43が神経機能を障害している。
Mitochondrial dysfunction
小胞体(ER)での局在に加えて、TDP-43はミトコンドリア内部に局在をしている(図1b)。ALSでのTDP-43変異とミトコンドリア外膜との関連を報告している研究と矛盾しているが、野生型または変異型TDP-43はミトコンドリアをターゲットとしている。ミトコンドリアの単離に高濃度の6M KClを使用した研究では、脂質とタンパク質の間の静電力を破壊し、ミトコンドリアの統合性およびプロテオームを障害している。
ALS発症におけるミトコンドリア機能不全の役割が明らかになっている。ALS関連SOD1変異を発現した様々な細胞や動物モデルにおいて、ミトコンドリアの機能不全とミトコンドリア中のSOD1の蓄積を報告している。ミトコンドリアの機能不全もまた、FTLD関連タウを発現するトランスジェニックマウスで研究されている。興味深いことに、TDP-43がALSまたはFTLD患者の神経細胞のミトコンドリア内に蓄積している。 TDP-43はin vitroでミトコンドリア内に取り込まれ、核内局在の減少またはTDP-43のALS関連変異のいずれかがミトコンドリア内での局在化を増強している。いくつかのTDP-43モチーフの欠失は、そのミトコンドリア局在化を抑制したが、完全ではなかったことから、ミトコンドリアへのTDP-43の局在化には複数の配列が必要である可能性があることが示唆された。ミトコンドリア内では、TDP-43は、ミトコンドリア転写mRNAまたはトランスファーRNA(tRNA)のサブセットに結合して、酸化的リン酸化複合体1の機能を特異的に損なう。TDP-43ミトコンドリア局在抑制は、ALSまたはFTLDのモデルマウスであるTDP-43トランスジェニックマウスにおいて、TDP-43誘発ミトコンドリア機能不全を抑制した。したがって、細胞質TDP-43封入体は、ミトコンドリアを標的にして、ALSやFTLDで神経毒性を引き起こす可能性がある。別の研究では、モデルマウスが顕著な神経細胞死、ミトコンドリアのクリスタ消失、およびミトコンドリア輸送障害を示すにもかかわらず、変異型TDP-43トランスジェニックマウスにおいてミトコンドリア機能が変化しないことを報告している。ADにおけるTDP-43ミトコンドリア局在およびそのミトコンドリア機能との関係についての研究は、これまで報告されていない。しかし、ALSと同様に、ミトコンドリア機能障害はADの顕著で初期の特徴であり、複合体Iの機能障害はADでは以前から報告されている。興味深いことに、TDP-43はミトコンドリア転写RNAと結合し、酸化的リン酸化複合体1の活性を調節する。ミトコンドリアは、Aβおよびタウの標的として注目されており、ミトコンドリアの機能を障害する点で、Aβ、タウおよびTDP-43の病原性経路が集約する可能性がある。
全体的に、いくつかの矛盾があるが、野生型または変異型TDP-43を発現する細胞またはニューロンにおいて一貫して報告されているミトコンドリア機能不全は、軸索輸送障害に加えて、細胞質内TDP-43蓄積がミトコンドリアの生物エネルギーに直接干渉して神経細胞の機能不全および喪失を生じさせる。ミトコンドリアの生物エネルギーにおけるAβ、タウ、およびTDP-43の機能的相互作用のさらなる詳細な調査が必要とされる。
Protein quality control
UBQLN2、p62、VCP、optineurinなどの多くのALSまたはFTLD関連タンパク質は、タンパク質の品質管理システムに関与している。p62は、ユビキチンプロテアソームシステム(UPS)とオートファジーを結びつけている。また、FTLD-TDPの大脳皮質において、TDP-43とp62の間の物理的相互作用の障害が報告されている。これらの知見は、TDP-43 proteinopathyと関連する神経変性が、系統的な蛋白質の品質管理の障害が推測される。ユビキチンプロテアソームシステム(UPS)またはオートファジー経路のいずれかを抑制すると、TDP-43細胞質蓄積および神経毒性を引き起こす。脳神経細胞では、変異型TDP-43の過剰発現により、アンフォールドタンパク応答(アンフォールドされていない、または誤ってフォールドされたタンパクに反応し、小胞体内に蓄積する)、ユビキチン凝集およびゴルジ断片化が誘導され、神経細胞脱落に先立って起こる。しかし、一部のモデルマウスの運動ニューロンでは、オートファジー誘導因子Atg7ではなく、プロテアソームサブユニットRpt3の消失のみで、TDP-43の細胞質蓄積と核内小胞体を引き起こす。今後、より詳細な検討が必要である。
Post‐translational modifications
TDP-43封入体では、ユビキチン化、高リン酸化、異常切断などの異常な翻訳後修飾が起こっている。 TDP-43 のユビキチン化部位はまだほとんど解明されていないが、TDP-43 内にはセリン、スレオニン、チロシン残基などの多くのリン酸化部位が同定されており、その中でもセリン 403、404、409、410 のリン酸化部位が広く研究されている(図 1c)。in vitro実験では、セリン379、403/S404、および409/S410におけるTDP-43リン酸化に関与するキナーゼの1つとしてカゼインキナーゼを同定し、TDP-43リン酸化がそのオリゴマー化または凝集を制御する役割がある。ALSまたはFTLD や野生型または変異型TDP-43を発現する細胞モデルおよび動物モデルでは、分子量20~25 kDaまたは35 kDaのTDP-43 N末端断片(NTF)または分子量20~25 kDaまたは35 kDaのCTD断片が報告されている。これまでの研究では、TDP-43の切断を媒介するカルパイン、カスパーゼ3、カスパーゼ7などのいくつかのプロテアーゼが知られている。ALS/FTLDに関連するほぼすべての変異は、TDP-43のCTDに見られる。NTFはそのスプライシング活性に必要である。CTD領域は、hnRNP相互作用およびmRNAスプライシング活性に必須であり、一般に、細胞質TDP-43封入体の形成に関与していると考えられている。CTDにおける331-369アミノ酸のQNリッチ領域は、アミロイド様βシート構造を形成することが報告されている。神経毒性の引き金となるTDP-43凝集体の病理学的役割はまだ不明であるが、病理学的なTDP-43の種が不溶性の凝集体を形成してニューロンに毒性を及ぼすことが示唆されている。細胞質TDP-43の蓄積の分子基盤も完全には解明されていないが、異常な翻訳後修飾、またはプリオン様ドメインまたはアミロイドを生じやすいドメインの存在が、TDP-43の誤まった局在化および凝集を媒介することが示唆されている。
Exosomes
ALSおよびFTLDの脳または脳脊髄液(CSF)から抽出された不溶性TDP-43凝集体は、TDP-43のリン酸化およびユビキチン化を誘導し、リン酸化およびユビキチン化されたTDP-43の凝集体を核化することが報告されている。さらに、いくつかの最近の研究は、エクソソームとして知られる分泌小胞を介してTDP-43が細胞から放出され、プリオン様TDP-43 凝集体の1つの細胞から別の細胞への拡散を促進し得ることを実証している(図1b)。これに沿って、GW4869による中性スフィンゴミエリン酵素2の不活性化によるエキソソーム分泌の阻害は、ヒトTDP-43 A315T変異体を発現するトランスジェニックマウスの病態を悪化させた。今後の研究では、細胞質TDP-43封入体がエクソソームを介したプリオン様拡散モデルを用いて、疾患の進行中に異なる脳領域に影響を与えるかどうか、またどのように影響を与えるかを検証する必要がある。
TDP‐43 in other neurodegenerative diseases
TDP-43 proteinopathyは、PDやHDなど他の多くの神経変性疾患においても報告されている。PDは、黒質のドーパミン作動性ニューロンを主に侵す神経変性疾患である。MNDを有するPD患者では、老人斑と局在的な神経現線維変化と共に、脊髄と球神経核にTDP-43陽性細胞質内封入体を認める(歯状回と大脳新皮質には存在しない)。また、TDP-43陽性封入体は、Parkin変異体を有する家族性PD患者のCA1における神経細胞脱落と関連していることが報告されている。パーキンの過剰発現は、細胞モデルおよび動物モデルにおいて、TDP-43誘導細胞死を緩和する。TDP-43の共発現は、変異型α-シヌクレインを発現するトランスジェニックマウスにおいてドーパミン作動性ニューロン脱落を悪化させ、PDにおけるTDP-43の相乗的役割の可能性をさらに示唆している。
TDP-43陽性封入体は、HD患者の多くの中枢神経系領域においてhuntingtin (Htt) 陽性封入体と共存しているとの報告がある。TDP-43が本当に封入体を持つHttと共局在するかどうかは議論の余地があるが、トランスジェニック線虫モデルにおいて、線虫のTDP-43相同分子を排除すると、Htt変異によって誘発された神経変性および行動異常が減少することが示されており、Htt変異の毒性を誘導するTDP-43の機能解析が必要である。
TDP‐43 as a common therapeutic target for multiple neurodegenerative diseases
種々の神経変性疾患におけるTDP‐43 proteinopathyの存在は、TDP‐43の病態生理に注視することで、これらの疾患を治療するための共通の治療法が解明される可能性がある。ストレス顆粒形成は、タンパク質凝集の初期段階として示唆されている。ataxin‐2がTDP-43の取込を介したストレス顆粒の形成に重要であることが報告されている。TDP-43の遺伝子変異は、ALS/FTLDのごく一部に過ぎない。変異型SOD1を標的としたアンチセンスオリゴヌクレオチド(ASO)が基盤の治療法とは異なり、変異型TDP-43を標的としたASOは、あまり意義を持たないかもしれない。しかしながら、Ataxin-2を標的としたASOベースの治療は、最近、ALS関連変異TDP-43を発現するトランスジェニック動物モデルにおいて、TDP-43とAtaxin-2の相互作用をブロックし、TDP-43誘発神経毒性を緩和し、運動機能を改善し、生存期間を延長することが報告されている。TDP-43封入体形成の防止は、TDP-43関連神経変性疾患に対する有望な共通の治療アプローチである可能性があることを示唆している。
TDP-43由来の阻害ペプチド(PM1)によるTDP-43ミトコンドリア局在化の抑制が、運動ニューロンおよび皮質ニューロンの脱落を減少させ、ALSおよびFTLD様表現型を示す2つの変異TDP-43トランスジェニックマウスモデルにおいて、行動異常を予防し、さらには回復させることを示しており、TDP-43ミトコンドリア局在化の標的化は、有望な治療アプローチとして期待される。さらに、ラパマイシン、フルフェナジンジヒドロクロライド(FPZ)、メトトリメプラジン(MTM)および10-(4′-(N-ジエチルアミノ)ブチル)-2-クロロフェノキサジン(NCP)などのオートファジー活性化剤は、TDP-43実験モデルにおいて神経細胞を保護し、認知機能を改善することが報告されている。同様に、プロテインキナーゼR(PKR)様小胞体キナーゼ(PERK)およびelF2αリン酸化の低分子阻害剤であるGSK2606414によるelF2αシグナル伝達経路の阻害は、ハエにおけるTDP-43が誘発する登坂障害を緩和することが報告されている。上述したように、TDP-43はタンパク質の品質管理システムと関連している。したがって、TDP-43に関連する神経変性もまた、タンパク質クリアランスの強化またはタンパク質翻訳の減少のいずれかを標的とする治療的アプローチによって緩和される可能性がある。
Conclusions and perspectives
TDP-43 proteinopathy は、ALS、FTLD、AD、PD、HD、海馬硬化症、脊髄小脳失調症2型、およびアレクサンダー病を含む神経変性疾患における病理学的特徴として報告されている。病理学的および機序学的な証拠の増加は、TDP-43 proteinopathyが、様々な神経変性疾患の神経細胞脱落の根底にある収束的病理学的メカニズムである可能性を示唆している。TDP-43 proteinopathyが「 gain of toxicity 」または「 loss of function」を介して機能するのかどうかはまだ議論の余地があるが、疾患におけるTDP-43または変異型TDP-43の病態メカニズムは、当初提案されていたよりも複雑なものである。TDP-43の核内消失と細胞質内蓄積は、一見無関係な2つの現象であるが、RNA生合成、タンパク質凝集、軸索輸送、UPS、オートファジー、さらにはミトコンドリアのバイオエネルギーにまで関与する異なる病態と関連している。今後の研究では、様々な主要な神経変性疾患におけるTDP-43制御の生物学的・病理学的プロセスを評価・比較することが重要である。特筆すべきは、TDP-43 proteinopathy だけでは神経変性を誘導できない可能性がある。TDP-43 proteinopathyと他の多くの病理学的特徴を考えると、TDP-43は他の因子と協力して神経変性を誘発する可能性がある。この点からも、他の病態との関連でTDP-43の病態メカニズムを解明することは、特に重要である。また、Aβ、タウ、その他多くの神経変性関連因子の病態メカニズムがTDP-43にも当てはまるかどうかを検討することも重要である。
過去10年間で、神経変性におけるTDP-43の病態整理についての理解が深まっていることから、TDP-43の翻訳研究を前進させることが急務であることが浮き彫りになってきた。TDP-43が介在するRNAスプライシングは、TDP-43の特定のRNAターゲットを予後のバイオマーカーとしての可能性がある。ストレス顆粒、ミトコンドリア、オートファジー、およびタンパク質合成を標的としたTDP-43の研究は、これらのASO、ペプチド、および低分子を第I相臨床試験に移行させるための実現可能性をサポートしている。様々な神経変性疾患で増加しているTDP-43 proteinopathyの原因は多因子性である可能性がある。最近の研究では、TDP-43を標的とすることが神経変性を予防するための一般的な治療法である可能性が示唆され、TDP-43の病態メカニズムに関する知見を臨床応用するためには、さらなる努力が必要である。
参考文献:Ju Gao, Luwen Wang, Mikayla L Huntley, George Perry, Xinglong Wang. Pathomechanisms of TDP‐43 in neurodegeneration. J Neurochem. 2018 Feb 27;10.1111/jnc.14327. doi: 10.1111/jnc.14327.