Synucleinopathies
2017年は、ジェームズ・パーキンソンが彼の名を冠した疾患の最初の完全な臨床記述を提供してから約200年、高用量D,L-DOPA治療が導入されてから約50年、α-シヌクレイン凝集が登場してから約20年である。1998年には、パーキンソン病とレビー小体型認知症に加わり、多系統萎縮症がが第三のsynucleinopathyとなった。ここでは、Lewy bodies、Lewy neurites、Glial cytoplasmic inclusions (Papp-Lantos bodies) におけるαシヌクレインの同定につながった研究と、それ以降の論文をレビューする。
はじめに
パーキンソン病(PD)の歴史は長い。一部の古文書には部分的な記述が見られるが、この病気の最初の完全な臨床的記述は1817年にJames Parkinsonによってなされ、この疾患の特徴的な運動症状、不随意の震え、筋力低下、前傾姿勢、歩行から走行への変化(突進現象)を記載している。。ジャン=マルタン・シャルコーはパーキンソンにちなんで "paralysie agitante"と命名している。
黒質緻密帯の色素神経細胞脱落と中脳や他の脳幹領域に存在するLewy bodiesが、PDの病理学的病変として同定されるまでに、100年近くの歳月を要した。Friedrich Lewyは最初にPD症例の迷走神経背側運動核、Meynert基底核、視床核にLewy bodiesを記述している。Rolf Hasslerは、PDにおける神経細胞脱落は腹外側の黒質で最も重篤であり、色素細胞密度の高い背内側神経細胞群は相対的に保持されていることを示した。1960年代に電子顕微鏡像にてLewy bodiesが異常なフィラメントで形成されていることが明らかになった。脳神経伝達物質としてのドーパミンの発見と、ドーパミンの80%が線条体に存在し、ノルアドレナリンを伴わないという発見と、レセルピンが脳内ドーパミンを枯渇させ、レボドパがドーパミンレベルを上昇させることを示す実験が相まって、Arvid Carlssonはドーパミン欠乏がPDの運動症状の根底にあるのではないかと提案した。その後、Herbert EhringerとOleh Hornykiewiczによって、パーキンソン病の6例(4例は脳症後パーキンソン病、2例はPD)の線条体ドーパミン欠乏が記録された。ドーパミンは黒質でも減少していることが明らかになった。Falck-Hillarp蛍光組織化学的手法の出現により、ドーパミンを含む神経細胞体が黒質緻密帯で発見され、そこから神経末端が線条体へと投射することがわかった。サルを用いた実験では、黒質病変に続いて線条体のドーパミンが消失することが示された。
このため、減少したドーパミンを補充する試みが行われた。カテコラミンは血液脳関門を越えることができない。そのため、血液脳関門を通過し、芳香族アミノ酸デカルボキシラーゼによってドーパミンに脱炭酸されるドーパミン前駆体3,4-ジヒドロキシフェニルアラニン(DOPA)が投与された。結局、パーキンソンの病気の記述から、高用量経口レボドパ治療が開発されるまでに150年かかった。1967年の論文では、D-L-DOPAが使用されたが、D異性体は生体内ではドーパミンに変換されないため、1969年の研究ではDOPAのL異性体が使用された。1970年、L-DOPAはPDの治療薬として米国食品医薬品局によって承認された。
Cotziasらは、PDは神経メラニン(ドパミンの最終産物)の減少が原因であると考えていた。彼らは大量のレボドパを投与し、枯渇したドーパミンを補充するのではなく、黒質の再色素化を試みた。その結果、PDの症状はかなり改善した。彼らの場合は、間違った理由で正しい結果を得たケースであったようである。現在、PDに対するメカニズムに基づいた治療法はない。レボドパの奇跡的な効果は、オリバー・サックスの脳後パーキンソン病患者の治療に関する記述である「Awakenings」に記載されている。彼の著書は、ハロルド・ピンターの戯曲「アラスカの一種」や、ペニー・マーシャル監督の映画「Awakenings」に影響を与えた。
これらの進歩にもかかわらず、PDの原因は不明のままであった。1-メチル-4-フェニル-1,2,5,6-テトラヒドロピリジン(MPTP)の注射後に複数の患者で突然発症したパーキンソニズムの報告により、環境因子が模索され、PDはほとんどが散発性の障害であると考えられていた。David MarsdenはPDをレビー小体型疾患と表現し、「パーキンソン病の核心はレビー小体型であるが、この封入体の起源は謎である」と書いている。Lewy bodiesやLewy neuritesには免疫組織化学的に多数のタンパク質が報告されていたが、この研究ではLewy bodiesの本質的な成分と、異常なフィラメントの中に閉じ込められる細胞成分を区別することはできなかった。
アルツハイマー病(AD)やPDを含む最も一般的な神経変性疾患を特徴づける封入体の組成を生化学、構造生物学、分子生物学を用いて解明することが可能であり、それによってそれらの病因や病態に光を当てることができると考えられていた。偶然にも、これはADの場合にも当てはまったが、PDのLewy bodies病理を解明するためには、遺伝学、生化学、構造生物学、分子生物学を結合させなければならなかった。
ヒトα-synuclein と β-synuclein
synucleinopathiesの発見は、ADの対のpaired helical filamentsのタウに関する研究から発展した。1990年代初頭、抗タウ抗体11.57が、正常な成人ヒト脳の細胞質抽出物から得られた2つのタンパク質が標識されていることが注目された(各タンパク質の見かけの分子量は19 kDa)。部分的な精製は、両方のタンパク質の熱安定性とイオン交換クロマトグラフィーによる分離を利用した。完全な精製は、追加のHPLCステップを使用した。エドマン分解で、アミノ末端分析は出来なかった(アセチル化、フォルミル化?)が、臭化シアン切断後に精製されたペプチドの分析は、自動アミノ酸配列解析によって55個の連続残基まで配列を決定することを可能にした。これらの成功した結果から、当初、2つのタンパク質を「Perfectin」(α-シヌクレイン)および「Imperfectin」(β-シヌクレイン)と命名した。次に、ポリメラーゼ連鎖反応(PCR)産物を用いて、成体ヒト海馬ライブラリーから完全長cDNAクローンを単離した。予測されたタンパク質は、140(Perfectin)および134(Imperfectin)アミノ酸から構成されていた。
1993年11月16-19日に大磯で開催された「第2回認知症国際シンポジウム」で斉藤綱男氏は、ADアミロイドの非Aβ成分(NACP)の前駆体が140アミノ酸のタンパク質であり、NACP(ペプチドNAC)の残基61~95がADのAβ老人斑に不可欠な成分であると発表した。しかし、追加の抗体を使用したその後の研究は、これらの知見を確認することができず、NAC/NACPはAβ老人斑の不可欠な構成要素ではないことが明らかになった。NACPとの同一性に加えて、140アミノ酸のタンパク質は、太平洋電気エイ(Torpedo californica)やラット脳のシヌクレインと相同性があることがわかり、ヒトのシヌクレインであることが判明した。また、ヒト脳由来の134アミノ酸タンパク質がウシのphosphoneuroprotein-14と相同性があることも判明した。後者は、シナプスには存在するがグリア細胞には存在しない新しい脳特異的なタンパク質として同定されていた。
これらのタンパク質の間のこれまで知られていなかった類似性から、ヒト脳シヌクレインのファミリーの存在が確立された。140および134アミノ酸タンパク質をそれぞれα-シヌクレインおよびβ-シヌクレインと命名されている(140アミノ酸タンパク質は、陽イオン交換HPLCカラムから134アミノ酸タンパク質よりも先に溶出したため、α-シヌクレインと命名された)。配列決定により、β-シヌクレインはα-シヌクレインと61%同一であった(図2)。その後、127アミノ酸のγ-シヌクレインが同定された。シヌクレインは、高度に保存されたアミノ末端リピート領域、疎水性の中間領域、およびあまり保存されていない負に帯電したカルボキシ末端領域を有する。初期の研究では、NACP/α-シヌクレインはnatively unfolded proteinであることが報告されている。

図 ヒトα-シヌクレインとβ-シヌクレインの配列比較。
α-シヌクレイン配列の半分以上は、11アミノ酸の7つの不完全なリピートによって占められており、それぞれが共通塩基配列KTKEGV(残基7~87)と保存された6アミノ酸のコアを持っている。個々のリピートのコア配列は、9個のアミノ酸で区切られているリピート4と5を除いて、5個のアミノ酸で区切られている。α-またはβ-シヌクレインのいずれかに特異的な抗体により、両タンパク質が、脳に優位に発現し、神経末端に集中していた。SNCA(α-シヌクレイン遺伝子)とSNCB(β-シヌクレイン遺伝子)は、それぞれ4番染色体(領域q21)と5番染色体(領域q35)の長腕に位置する遺伝子によってコードされている。また、γ-シヌクレイン遺伝子(SNCG)は、10番染色体(領域q21)の長腕にマップしている。PDの病因に関する多くの研究は環境危険因子の探索に焦点が当てられていたが、PDの遺伝例は知られていた。1990年に、早期発症のPDと剖検で確認されたレビー病理を有するイタリア系アメリカ人家族(Contursi家系)が報告された。1996年、Robert Nussbaumらは、この家族の第4染色体の長腕(q21-q23領域)にある遺伝マーカーを同定した。1997年6月、Mihael Polymeropoulosらは、Contursi家系で変異している遺伝子としてSNCAを同定し、140アミノ酸タンパク質の53位のアラニン-スレオニン置換の突然変異が存在した。この変異はイタリア系アメリカ人の親族だけでなく、血縁関係のないギリシャ系の3つの家族にも見られた。遺伝のパターンは常染色体優性であった。驚くべきことに、A53はヒトと旧世界霊長類のαシヌクレインにのみ存在し、T53はげっ歯類と他のいくつかの哺乳類種に存在している。
1997年8月、孤発性PD患者6名(図3)とレビー小体認知症認知症患者4名(DLB)の黒質のLewy bodies と Lewy neuritesがα-シヌクレインに対して強い免疫反応性を示した。DLBの帯状回皮質のLewy bodiesも同様であった。α-シヌクレインのアミノ末端およびカルボキシ末端に特異的な2種類の抗α-シヌクレイン抗体は、Lewy bodiesと反応した。β-シヌクレインに特異的な抗体はPDとDLBのLewy bodiesを染色しなかった。これらの所見から、脳幹型および皮質型Lewy bodiesはα-シヌクレインに対して免疫反応性であることが示された。PDとDLBの両方がαシヌクレイン疾患であると考えられた。αシヌクレイン封入体は、SNCA変異によって引き起こされる疾患の特徴でもある。
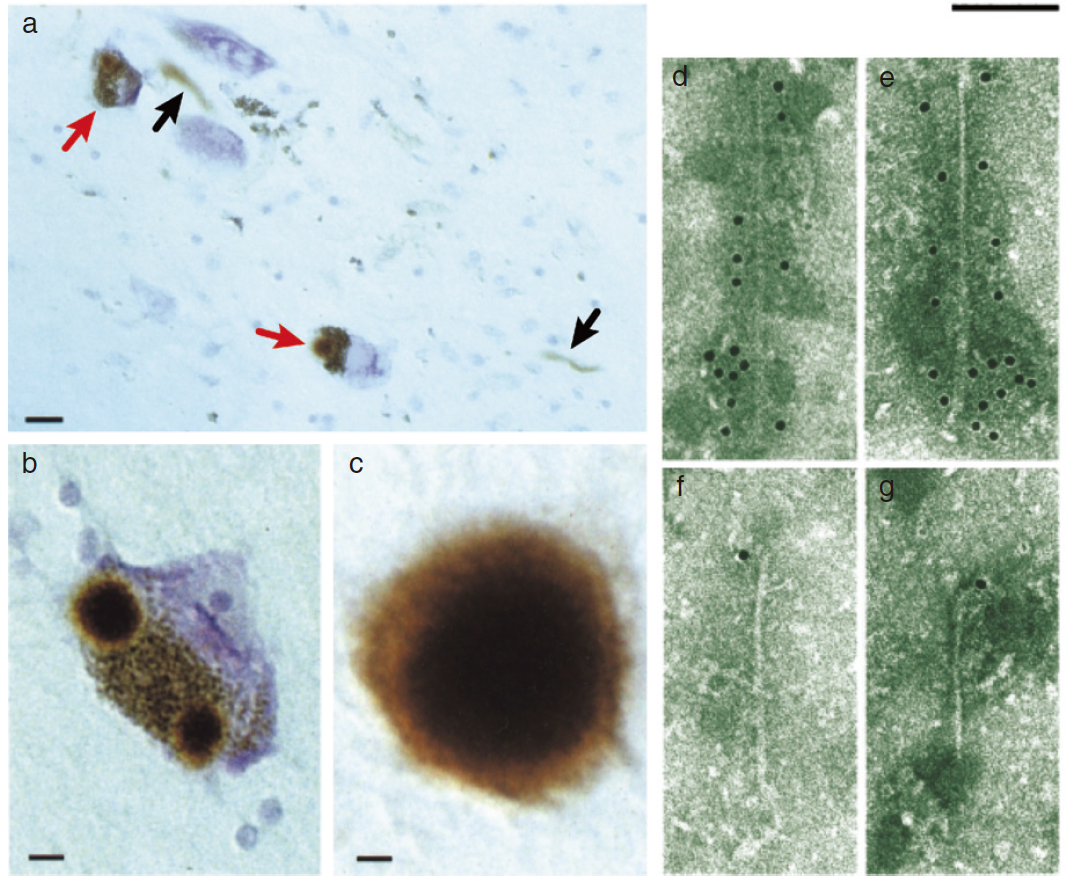
図 パーキンソン病のαシヌクレイン病理。黒質をはじめとする脳領域のLewy bodiesと Lewy neuritesは、神経病理学的にパーキンソン病を診断する。これらは、α-シヌクレイン抗体によって標識された光(a-c)および電子顕微鏡(d-g)レベルで示されている。(a)、α-シヌクレイン陽性のLewy bodies(赤矢印)を含む2つの色素性神経細胞。Lewy neurites(黒矢印)もα-シヌクレイン陽性である。スケールバー、20μm。(b)、αシヌクレイン陽性Lewy bodiesを2つ持つ色素性神経細胞。スケールバー、8μm。c)、α-シヌクレイン陽性の細胞外Lewy bodies。スケールバー、4μm。d-g)、パーキンソン病患者の黒質から単離されたフィラメントは、α-シヌクレインのカルボキシ末端(dおよびe)とアミノ末端(f,g)領域に金粒子が観察される。スケールバー、100 nm。
1998年5月、PDおよびDLBにおいてLewy neuritesがこれまで信じられていたよりも多く存在することが報告された(図4)。神経突起内Lewy bodiesの染色は、レビー病変が良性である可能性が低いという見解をサポートしている。これ以前は、ユビキチン染色がレビー病変を検出する最も感度の高い手段であったが、特異性に欠けるものであった。我々は、α-シヌクレインの染色がユビキチンの染色よりも広範囲であることを示し、α-シヌクレインの蓄積がユビキチン化に先行していることを示した。これらの結果から、α-シヌクレインのユビキチン化は線維形成後に起こり、プロテアソームによる分解のためにα-シヌクレインを標的にすることにより、凝集したα-シヌクレインのクリアランスを促進する細胞の試みである可能性が示唆された。
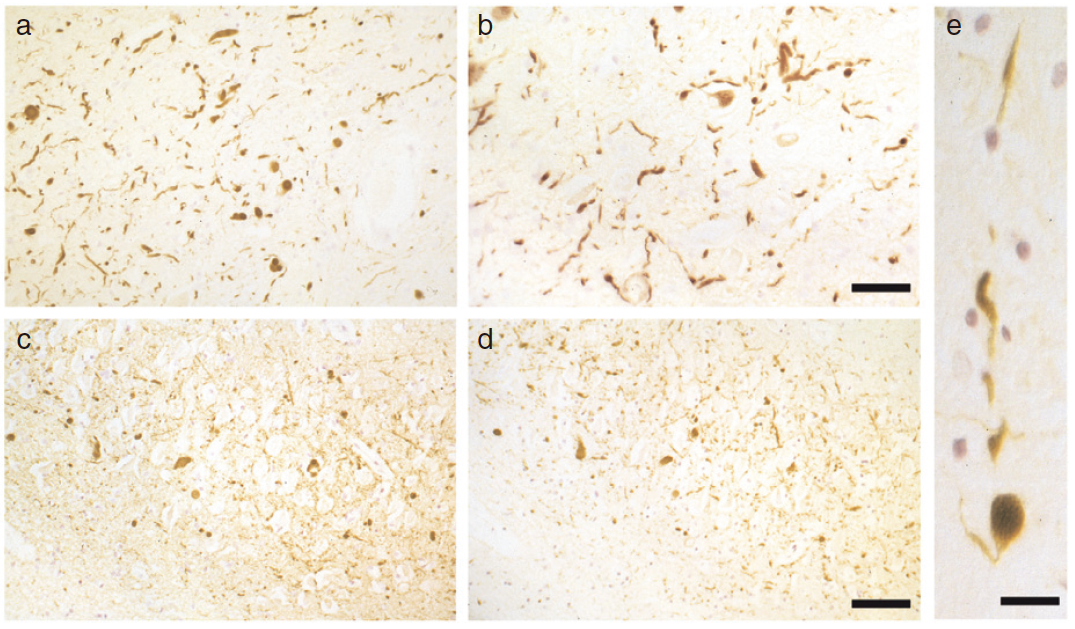
図 レビー小体認知症のαシヌクレイン病理.(a,b) α-シヌクレイン陽性のLewy bodiesと黒質部のLewy neurites。スケールバー、100μm。(c,d) 海馬のα-シヌクレイン陽性Lewy bodiesとLewy neurites。スケールバー、80μm。(e) α-シヌクレイン陽性Lewy bodiesと黒質突起のLewy neurites。スケールバー、40μm。
また、レビー病変ではβシヌクレインが蓄積しないことを確認し、γシヌクレインについても同様であることを示した。哺乳類の3つのシヌクレインのうち、レビー病変にはα-シヌクレインのみが存在する。これらの知見は、α-シヌクレインがLewy bodiesやLewy neuritesを構成する異常なフィラメントの主要な構成要素であることを示唆している。Tony Crowther、長谷川正人と共同で、DLB患者の帯状回皮質から抽出したサルコシル不溶性フィラメントを免疫電子顕微鏡で調べたところ、α-シヌクレインのカルボキシ末端領域を認識する抗体PER4は、直径5~10nm、長さ200~600nmのフィラメントを標識し、α-シヌクレインを主成分とすることを示した。一方、α-シヌクレインの残基 11-34 に対して上昇させた抗体 PER1 は、各フィラメントの一端のみを標識していた(図 3)。このことから、PER1エピトープはフィラメント本体に埋もれており、フィラメントは極性構造であることが示唆された。その後、PD患者の黒質から抽出したフィラメントについても同様であった。α-シヌクレインの染色は、現在では組織切片やPDやDLBにおけるレビー小体やレビー神経突起の同定に日常的に用いられている。
α-シヌクレインとオリゴデングリア細胞質封入体(Papp-Lantos体)
多系統萎縮症(MSA)は、自律神経症状、小脳症状、パーキンソン症状、錐体症状によって特徴づけられる神経変性疾患である。運動表現型により、パーキンソン病型(MSA-P)と小脳型(MSA-C)に分けられる。MSA-PはL-DOPA反応性が悪い。MSAは、かつてオリーブ橋小脳萎縮症、線条体黒質変性症、シャイ・ドレーガー症候群と呼ばれていた。ほとんどの国では、MSA-CよりもMSA-Pの方が多い。PDと同様に、MSAの発症は通常、60歳台であるが、PDよりも進行が早く、発症からの平均生存期間はわずか6~10年である。
タンパク質性オリゴデングリア細胞質封入体(Papp-Lantos体)は、MSAの主要な組織学的特徴である。オリゴデンドログリア核封入体、神経細胞の細胞質内封入体および核内封入体もまれであるが観察される。シュワン細胞細胞質内封入体も一般的な所見である。
1998 年 7 月に MSA のグリアとニューロンの封入体はα-シヌクレインのアミノ末端とカルボキシ末端を認識する抗体で染色され、 α-シヌクレインが含まれていることが明らかになった(図 5)。二重染色により、α-シヌクレインの染色はユビキチンの染色よりも広範囲であることが示され、α-シヌクレインの蓄積がユビキチン化に先行していることが示唆された。β-またはγ-シヌクレインに対する抗体では、グリアまたはニューロンの封入体は染色されなかった。
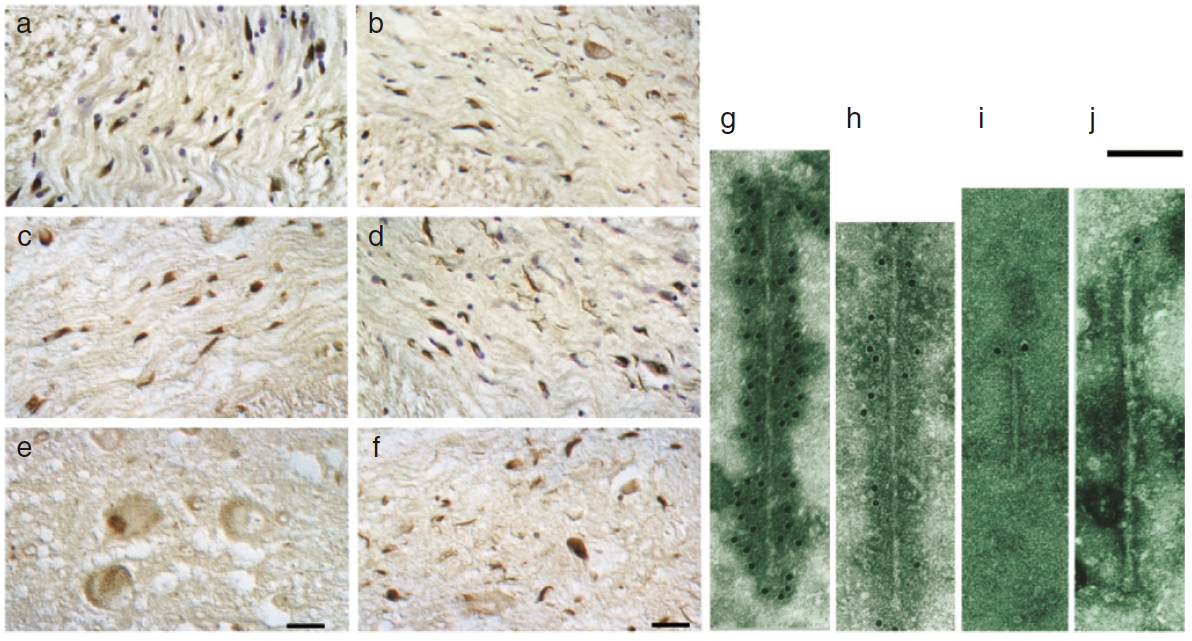
図 多系統萎縮症のαシヌクレイン病理 脳内のグリア細胞質内封入体の存在は、多系統萎縮を確定する。同様の封入体は、グリア細胞の核、および神経細胞の細胞質や突起内、核内に存在する。光顕レベル(a-f)と電子顕微鏡レベル(g-j)で、封入体は、α-シヌクレイン抗体で標識される。(a-d)橋(a,b,d)と小脳(c,e,f)の白質におけるα-シヌクレイン陽性オリゴデンドロサイトと神経細胞。橋(e)と前頭葉大脳皮質(f)の灰白質におけるα-シヌクレイン陽性オリゴデンドロサイトと神経細胞。矢印は特徴的な病変を示す:細胞質内オリゴデンドログリア封入体(a,f)、神経細胞細胞質内封入体(b)、オリゴデンドログリア核内封入体(c)、 neuropil threads(d)、神経細胞核内封入体(e)。スケールバー、(e)で33μm、(f)で50μm。(g-j), 多系統萎縮症例の前頭葉大脳皮質および小脳から単離されたフィラメントは、α-シヌクレインのカルボキシ末端(g,h)およびアミノ末端(i,j)領域に特異的な抗体により標識(金粒子)されている。(g,h)の一様な標識と(i,j)のフィラメント末端のみの標識に注意。(g)はtwisted filamentを示しており、(h)はstraight filamentを示す。スケールバー、100 nm。
MSA 脳から抽出単離されたサルコシル不溶性フィラメントは、直径が 5~18 nm であり、カルボキシ末端領域を認識する抗体PER4 によって強く標識されていた。アミノ末端領域を認識する抗体PER1は、PDおよびDLBの場合と同様に、フィラメントの片方の端のみを標識していた(図5)。α-シヌクレインフィラメントが主に神経細胞の細胞質にレビー小体やレビー神経突起の形で存在するPDやDLBとは異なり、MSAでは、α-シヌクレイン陽性の介在物が神経細胞とグリア細胞の両方の細胞質や核に存在する。1998年以来、PD、DLBおよびMSAはしばしば synucleinopathies と呼ばれている。
その後の20年
synucleinopathiesはヒト神経変性疾患の新しいグループとして、PD、DLB、およびMSAを含む。レビー小体病は、末梢神経系にレビー病理を認める純粋自律神経障害病を含む疾患である。偶発的レビー小体病(incidental Lewy body disease)では、60歳以上の患者の5~10%に存在し、PDの前臨床型である可能性がある。臨床症状がなくてもLewy bodiesおよびLewy neuritesが存在する。パーキンソニズムなど臨床症状発現時には、黒質のドーパミン作動性ニューロンの約30%、軸索末端の50~60%が消失している。偶発的レビー小体型病では、黒質緻密帯神経細胞脱落は研究により有意差は異なり、結論は出ていない。
α-シヌクレイン封入体は、他の神経変性疾患でも見られる。このように、遺伝性および孤発性ADでは、約60%がレビー病理を持ち、特に扁桃体に存在している。しかしながら、パーキンソニズムのすべての症例がα-シヌクレイン封入体の存在があるわけではなく、脳炎後パーキンソニズムではアルツハイマー型タウ陽性NFTが認められる。
パーキンソニズムは、硬直、安静時振戦、歩行障害の3つの特徴のうち少なくとも1つを伴って、時間の経過とともに悪化する運動障害と定義される。PDの診断を確定することは生存中には不可能であり、75~95%の患者は剖検で診断が確定している。臨床的な定義には支持的所見が重要である;支持的所見には、L-DOPAに対する反応性、非対称性、および最近では非運動症状の存在が含まれる。後者は、前駆PDという概念を生み出している。
α-シヌクレインの生理
α-シヌクレインの生理機能は完全に理解されていない。α-シヌクレインは、アミノ末端リピートを介して酸性リン脂質と結合し、多量体化してα-ヘリカル構造になり、α-シヌクレインは膜を再構築する。ラット脳シナプスボタンには、平均300個のシナプス小胞に約3,500個のα-シヌクレイン分子が存在している。神経細胞ではα-シヌクレインは軸索末端に集中し、ミトコンドリアの多くは神経細胞体と樹状突起に局在しているが、ミトコンドリアはα-シヌクレインの発現により断片化することが判明している。α-シヌクレインが過剰発現するとミトコンドリアに局在する可能性がある。
SNCA欠失は神経変性をもたらさない。 α-シヌクレイン欠失マウスは線条体ドーパミンの放出量の増加を示した。逆に、α-シヌクレインを過剰発現させたマウスでは、神経伝達物質の放出が減少する。α-シヌクレインは神経伝達物質の放出を減衰させ、おそらくシナプスでの多量体化を介して、SNAREタンパク質の再分配とシナプス小胞のクラスタリングをもたらしていると考えられる。SNCA遺伝子座の欠失したC57BL/6Jマウスにおいても、α-シヌクレインのドーパミン作動性ニューロンに対する機能が残っている。
3つのシヌクレインの存在は、α-シヌクレインノックアウトマウスの比較的控えめな表現型に重複性があることを提起した。その後、α-、β-およびγ-シヌクレインを欠失したマウスが作製され、これらのマウスでは、シナプス小胞がシナプス前膜と融合する傾向が強まったためか、単独のノックアウトマウスよりも線条体ドーパミン放出量が増加した。しかし、いずれの表現型は比較的軽度の増加であった。線虫やショウジョウバエではシヌクレインの相同体は確認されていない。おそらく、α-シヌクレインの生体内での最も強い効果は、シナプス前シャペロンであるcysteine string protein(CSPα)欠失を補助する能力である。CSPαのノックアウトはマウスに、2ヶ月以内に進行性のシナプス変性と死をもたらす。α-シヌクレインの過剰発現はCSPαの欠損によるシナプス変性を大幅に遅らせ、α-シヌクレイン欠失はCSPαノックアウトによる影響を増悪させた。これらの知見は、α-シヌクレインがCSPαと同様にシャペロンである可能性を示唆している。しかし、α-シヌクレインがCSPα欠損を救うメカニズムは不明である。α-シヌクレインの脂質結合能が大幅に低下したA30Pα-シヌクレインでは、CSPα欠失に起因する神経変性を改善させないことから、酸性リン脂質の結合能が関係している可能性が考えられている。
α-シヌクレイン封入体
ヒト脳の病理学的封入体は、フィラメントのコアはα-シヌクレインの反復領域(残基30-110)を含む約70アミノ酸が伸長している。ヒトαシヌクレインの残基68-78の結晶構造は、各シートに平行なβ鎖を持つ対のβシートと、シート間に反平行なβ鎖を持つ対のβシートを示していた。対になったシート間の領域を示すzipper structureは他の構造に比べて長く、各対のβシートは2つの水分子を含んでいる。固体核磁気共鳴、走査型透過電子顕微鏡、X線回折によって得られた高分解能構造では、α-シヌクレインフィブリルのコア残基は、 Greek keyのトポロジーを持つ平行なin-register βシートに配置されていた。
凝集したαシヌクレインの90%以上はセリン129でリン酸化されているが、正常な脳のαシヌクレインのうち、この部位でリン酸化されているのは約4%である。リン酸化されたαシヌクレインが異常なフィラメントを形成するかどうかは不明である。セリン129でリン酸化されたα-シヌクレインを認識する抗体は、PD、DLBおよびMSAの介在物を同定するための感度の高い特異的なツールである。α-シヌクレインは、G蛋白質結合受容体キナーゼ、カゼインキナーゼおよびポロ様キナーゼを含む複数のタンパク質キナーゼによって、この部位でリン酸化され得る。組換え哺乳類α-シヌクレインは、ヒト脳のフィラメントの形態学的および超構造学的特徴の多くを共有する(図6)。α-シヌクレインが凝集すると、βシートに富んだ構造を持つようになる。凝集は、α-シヌクレインのアミノ末端100アミノ酸の配列を介して起こる。残基71-82を欠失すると、α-シヌクレインがフィラメントに凝集する能力がなくなる。同様に、残基66-74の欠失もまた、凝集を阻害した。対照的に、α-シヌクレインのカルボキシ末端領域は凝集を阻害した。
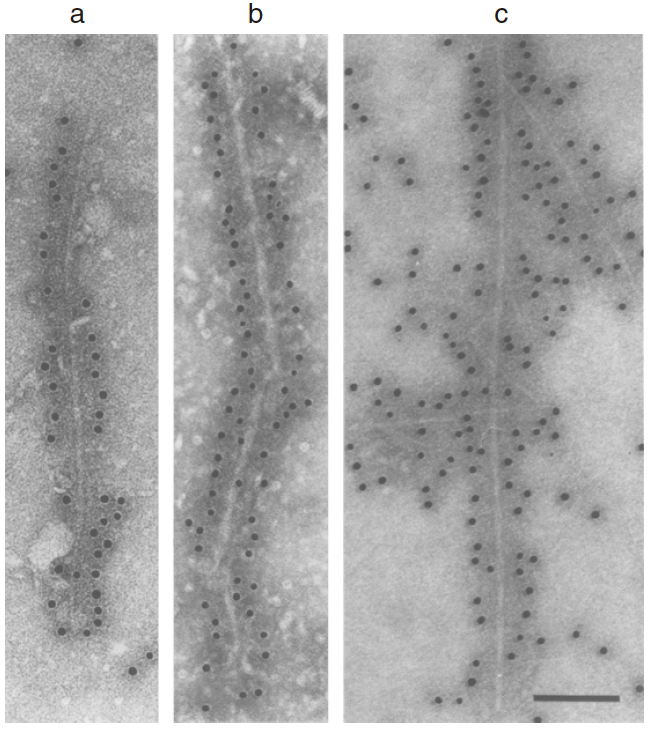
図 レビー小体認知症(a)と多系統萎縮症(b)から抽出されたフィラメント、組換えヒトαシヌクレイン(c)で組み立てられたフィラメントを抗αシヌクレイン抗体で標識。
ヒトのβ-シヌクレインとγ-シヌクレインはフィラメントへの凝集はなく、natively unfolded conformationのままであった。β-シヌクレインは、α-シヌクレインの残基73-83にまたがる疎水性領域を欠いており、この違いがタンパク質のフィラメントへの凝集性の違いがあることを示唆している。しかし、α-シヌクレインの残基73-83を欠失させても、フィラメントの凝集性は低下しなかった。さらに、α-シヌクレインの残基73~83が挿入されたβ-シヌクレインからなるタンパク質は、野生型α-シヌクレインよりも低いフィブリル化傾向を有していた。α-シヌクレインのフィブリル化傾向は、アミノ酸組成およびβ鎖の連続性によって決定され、凝集するβ-シヌクレインの変異体を産生することができた。
特発性PDはPD症例の90%以上を占めている。ヒトの正常脳と疾患脳の研究では、α-シヌクレイン封入体が脳のさまざまな部位の神経細胞に出現し、α-シヌクレイン沈着の6つのステージに分けられている。脳内の最初のα-シヌクレイン陽性構造は、嗅球および/または舌咽神経および迷走神経の背側運動核に発生する(第1期)。その後、扁桃体および黒質(第3期)に続いて、延髄および橋の被蓋(第2期)でレビー病変が発現する。PDの運動症状が現れ始めるのは第3期である。側頭葉大脳皮質に達する(第4期)では、病理像は悪化する。第5期および第6期には、 Lewy bodiesおよびLewy neuritesが大脳皮質に発現し、進行PDに関連した認知障害の多くを占めるようになる。大量のα-シヌクレイン陽性アストロサイトが第4~6期に出現する。
α-シヌクレイン封入体は、初期に腸管神経系に形成され、迷走神経を介して延髄迷走神経背側運動核に連続している。初期に末梢神経系にα-シヌクレイン封入体が形成されることもある。この病気がどのようにして広がるのか、そのメカニズムは不明であるが、腸管から、迷走神経を介して逆行性に脳に移動する可能性がある。これを裏付けるように、迷走神経の切除はPDのリスクを低下させることが報告されている。あるいは、封入体形成は迷走神経背側運動核で始まり、そこから脊髄および腸に遠心性に移動する可能性がある。腸におけるレビー病理の分布は、迷走神経背側運動核からの入力とパラレルである。この病期分類は、ほとんどのPD患者が運動機能障害の前に非運動症状を発症するという事実と一致している。便秘、低汗症、抑うつ、急速な眼球運動行動障害は、パーキンソン氏がすでに気づいていたことで、運動症状に何年も先行する可能性がある。それらの所見はレビー病理の全身分布と相関している。いくつかの研究は、α-シヌクレインの凝集がシナプスで始まるという見解を支持している。
同様の病期分類法はMSAにはない。しかしながら、incidental Lewy body diseaseに類似した前駆状態が報告されている 。α-シヌクレイン封入体を多量に認める非定型MSAは、線条脳変性症とオリーブ橋小脳変性症の存在下では臨床的にも病理学的にも前頭側頭葉変性症を呈するが、自律神経機能障害を伴わない。MSA-CとMSA-Pの他に、MSA-FTLDは第三の疾患形態である可能性がある。
SNCAの遺伝学
家族性PDの原因として、SNCAの6つの優性遺伝性ミスセンス変異が報告されている(図7)。A53Tの他に、A30P、E46K、H50Q、G51D、A53Eがある。疾患の発症年齢は家族内でも変動するが、突然変異G51D、A53E、およびA53Tは最も早い発症年齢を示す。実験的に、E46K、H50Q、およびA53Tはα-シヌクレイン封入体形成を増加させ、一方、A30P、G51D、およびA53Eはα-シヌクレインの凝集を減少させる。A30P、G51DおよびA53Eはまた、酸性リン脂質と相互作用する変異型α-シヌクレインの能力低下をもたらす。これらの所見は、α-シヌクレインの脂質結合と細胞毒性種への凝集との間に拮抗的な関係が存在することを示唆した他の研究と一致する。
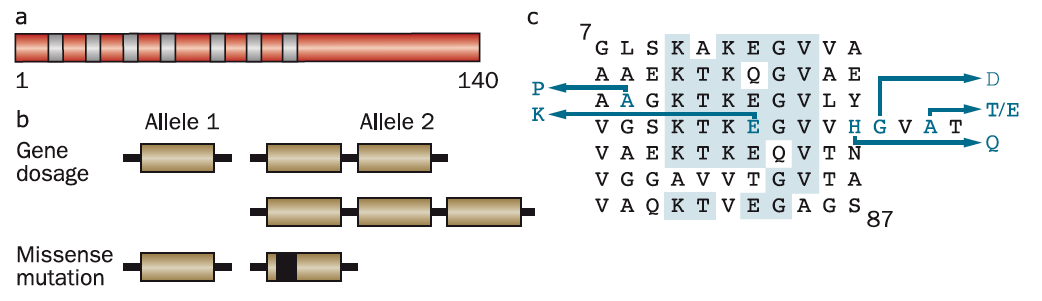
図 ヒトα-シヌクレインとその疾患責任遺伝子変異 (a)140アミノ酸のヒトαシヌクレイン蛋白質配列図。7つのアミノ末端リピートのコア領域を青い棒で示す。(b)SNCAを含む染色体領域の遺伝子量の増加(重複または三重化)またはSNCAミスセンス変異は、パーキンソン病やレビー小体を伴う認知症の優性遺伝型を引き起こす。(c)ヒトα-シヌクレインのリピート(残基7~87)を示し、病気の原因となるミスセンス変異 (A30P, E46K, H50Q, G51D, A53E と A53T) を青文字で示す。7つのリピートのうち、少なくとも5つのリピートで同一のアミノ酸は青字で示されている。
SNCAを構成する染色体領域の優勢遺伝的な重複および三重化もまた、PDを引き起こす。これらの場合、α-シヌクレインの配列は野生型であり、その特性の変化ではなくタンパク質レベルの上昇がPDを引き起こすことを示している。SNCAの複製は、発症年齢および症状の点で散発性障害と類似したPDの形態をもたらすが、三重化はより重度の表現型を引き起こし、発症年齢がより早く、顕著な認知障害を伴う。
特発性PDのリスクに関する遺伝子研究では、SNCAが最大の寄与をしていることが示された。示唆された多型はコーディング領域の外側に存在するため、おそらくmRNAの発現に影響を与え、結果としてα-シヌクレインの発現が増加する。LRRK2 (leucine-rich repeat kinase 2), GAK (cyclin G-associated kinase) とMAPT (microtubule-associated protein tau)の多型も関与する。SNCAおよびMAPTの多型は、MSAの危険因子である。
Synucleinopathiesの動物モデル
神経細胞にヒト野生型または変異型のα-シヌクレインを過剰発現させたトランスジェニックマウスでは、豊富なα-シヌクレインフィラメントが形成されることがある。封入体の形成は運動障害の発生と相関している。おそらく脳幹や脊髄の病理像が関連している。動物モデルとPDとの大きな違いは、黒質のドーパミン作動性神経細胞に有意な病理学的変化および神経変性が認められないことであった。
この問題は、αシヌクレイン欠損マウスにおいて、ラットチロシン水酸化酵素プロモーターの制御下でカルボキシ末端で切断されたヒトαシヌクレインを発現するトランスジェニックマウス系統によって部分的に解決されている。これらマウスでは、α-シヌクレイン凝集体、線条体ドーパミン欠乏症、および運動機能の低下を発症した。ヒトαシヌクレインのカルボキシ末端の切断は凝集を促進する。SNCAまたはSNCBノックアウトマウスで完全長のヒトα-シヌクレインを発現させた場合も凝集が促進される。野生型ヒトα-シヌクレインをオリゴデンドロサイトで発現させると、トランスジェニックマウスは脳および脊髄においてMSA様の変性を発症した。アストロサイトでA53T α-シヌクレインを誘導的に発現させたマウスでは、広範なアストロサイトーシス、マイクログリア活性化、神経細胞変性、および重度の麻痺が発現した。DLBをモデル化した神経細胞性シヌクレイン症の誘導性トランスジェニックマウスモデルでは、大脳辺縁系におけるα-シヌクレイン発現の抑制は、α-シヌクレイン封入体の除去とシナプス障害の消失をもたらした。
アデノ随伴型およびレンティウイルスベクターは、げっ歯類および霊長類の黒質においてヒト野生型および変異型α-シヌクレインを発現させるために使用されてきたが、Lewy bodies様封入体の形成および神経細胞の変性をもたらし、α-シヌクレインの凝集は神経変性を促進する。α-シヌクレインの膜結合は凝集を抑制し、一方、膜結合をブロックすると凝集が増強された。ベクリン1や転写因子EB(TFEB)の過剰発現を介したオートファジーの活性化は、α-シヌクレインの凝集を抑制した。興味深いことに、腹側被蓋領域(VTA)のドーパミン作動性ニューロンは、ウイルス性のA53Tαシヌクレインの効果に対して非常に抵抗性があり、TFEBを抑制すると変性した。PD で報告されているように、黒質と腹側被蓋領域のドーパミン神経ニューロンの脆弱性の違いは、リソソソームクリアランスの違いや酸化性リン酸化の酸化的リン酸化の高い基礎代謝率と軸索のより複雑な分枝によるものであろう。これは、1922年にCécileとOskar Vogtによって提唱された選択的脆弱性 (pathoclisis) の概念を彷彿とさせる。
ショウジョウバエにおけるヒトα-シヌクレインの発現は、糸状のレビー小体様封入体の形成、ドーパミン作動性ニューロン脱落、および運動障害をもたらした。神経変性では、α-シヌクレイン凝集がおこり、シャペロンが関与している。同様に、線虫のヒトαシヌクレインの過剰発現もまた、ドーパミン神経細胞脱落と運動障害をもたらす。
進行性PD患者の線条体に治療的に移植された胎児期のヒト中脳ニューロンにおけるレビー病変の発生からヒト脳におけるプリオン様メカニズムが考えられている。10年以上生存症例では、移植された細胞の2-5%にレビー病変が検出されたが、これはPDにおける黒質緻密帯にレビー病変を有するニューロンの割合とほぼ同じであった。24年後には、移植されたドーパミン作動性ニューロンの11~12%がα-シヌクレインおよびユビキチン陽性封入体を認めた。
過去9年間の実験研究により、動物へのα-シヌクレイン封入体の注入により、神経細胞が注入部位で細胞内封入体を形成し、そこから遠く離れた脳に広がり得ることが示されてきた。長期in vivoイメージングにより、凝集した組換えα-シヌクレインは、α-シヌクレイン陽性凝集体を発生することができた。封入体を含むニューロンは変性し、封入体形成が細胞毒性と関連していることを実証した。PD症例の黒質では、レビー小体型ニューロンの割合は約4%であり、時間が経過しても一定である。ニューロンが死滅すると封入体は排除されると考えられている。レビー病理によってニューロンが死滅するモデルでは、 Lewy bodiesの平均生存時間は6ヵ月と推定されている。
疾患に関連したα-シヌクレインフィラメントの形態学が報告されている。レビー病理はCampbell-Switzer鍍銀陽性、 Gallyas-Braak鍍銀陰性であった。対照的に、MSAのグリア細胞質封入体は、Campbell-Switzer鍍銀とGallyas-Braak鍍銀の両方で陽性であった。MSA患者からの脳抽出物は、PD患者からの脳抽出物とは対照的に、ヒトA53T α-シヌクレインに対するヘテロ接合体トランスジェニックマウスで増殖した。しかし、MSAとは異なり、α-シヌクレイン封入体は神経細胞に存在した。
組換えαシヌクレイン凝集は、リボンまたはフィブリルなど形態の多型が報告されている。ラット黒質に注入すると、リボン型はレビー病変を生じさせたが、フィブリル型はレビー病理を生ぜず、ドーパミン作動性ニューロン脱落をもたらした。リボンとフィブリルがヒトのシヌクレイン病に対応するものを持っているかどうかは、まだ不明である。いくつかのα-シヌクレインフィラメントはタウおよびα-シヌクレイン凝集の両方を生じたが、あるものはα-シヌクレイン凝集のみを生じた。α-シヌクレイン凝集構造物は、プロテイナーゼK消化後に異なる特性を示した。これらは、構造的変異、特性の違い、および表現型形質の遺伝性を示すという点で、プリオン種に類似していた。
誘導多能性幹細胞
SNCA変異キャリアからの誘導多能性幹細胞(iPSC)由来の神経細胞は、実験系としてますます重要な役割を果たしている。その主な利点は、それらがヒト神経細胞であるという事実にある。しかし、synucleinopathiesの場合のように、毒性機能の獲得メカニズムによって引き起こされると考えられている成人発症疾患のモデル化へのiPSC技術の応用は、依然として困難な状況にある。原理的には、疾患につながる初期の病理学的変化は、これらの系で研究することができるが、その正しい解釈には最終的に病理学的検索が必要となる。
結論
α-シヌクレイン異常凝集は、PD、DLB、MSAの「ロゼッタストーン」である。疾患の病因や病態を理解し、安全で効果的なメカニズムに基づいた治療法を開発することが必要である。PD、DLB、MSAに対するメカニズムに基づいた治療法が利用可能になれば、おそらくα-シヌクレインの凝集によって引き起こされる神経変性を阻害することになるであろう。前臨床段階が長いことを考えると、リスクのある患者を特定できれば、予防戦略を開発できる。原則として、疾患につながる初期の病理学的変化はモデル系で研究することができる。しかし、それらの正しい評価には、最終的に病理学的検索が必要となる。
参考文献:Goedert M, Jakes R, and Maria Grazia Spillantini MG. The Synucleinopathies: Twenty Years On. Journal of Parkinson’s Disease 7 (2017) S51–S69, DOI 10.3233/JPD-179005